COLLOQUE BOBIGNY 2017 résultats des études pivot en cancérologie sont-ils
CLIQUEZ SUR LE LIEN CI-DESSUS POUR VOIR LE DIAPORAMA
Les résultats précoces des études « pivot »[1] (nouveau terme pour essai clinique) en cancérologie sont-ils fiables au vu des publications à long terme de ces mêmes essais cliniques ? [2] [3]
Colloque_2017_bobigny-poster-SSP_G_DELEPINE colloque bobigny 2017 resultats-pivots
Résumé
Introduction.
Les études pivots (ou études cliniques ou encore essais thérapeutiques) sont celles qui sont soumises rapidement aux agences du médicament pour obtenir l’autorisation express de mise sur le marché (AMM) d’une nouvelle molécule. Afin de tester la fiabilité de leurs résultats rendus après quelques mois, nous les avons comparés aux données publiées tardivement pour ces mêmes études.
Matériel et méthodes.
Une recherche informatisée, avec pour mots clefs : Haute autorité de santé (HAS), Food and Drug Administration (FDA) , European Medication Agency (EMA) couplés à avastin, herceptine, erbitux, tarceva, nexavar, iressa, xalcori, giotrif, torisel, votrient, sutent, inlyta dans les cancers broncho-pulmonaires épidermoïdes, du rein, du colon, du sein et ORL, a été réalisé permettant de retrouver 42 études pivots correspondant à ces nouvelles molécules dites innovantes.
Nous avons ensuite recherché celles dont les résultats lointains précisant les durées de survie sans progression (= stabilisation tumorale), la survie globale et la toxicité ont été publiées (32), puis comparé les résultats des études pivot à ceux des derniers essais plus tardifs sur ces trois critères.
Résultats. Moins de 30% des résultats présentés aux agences de régulation, lors des demandes d’AMM, sont confirmés totalement par des essais ultérieurs. 20% d’entre eux sont confirmés partiellement. Les discordances constatées entre résultats initiaux et tardifs se fait toujours dans le sens d’une affirmation de plus grande efficacité de la molécule et de sa moindre toxicité de la lors de l’étude pivot, que d’après les résultats tardifs.
Conclusions. En matière de thérapies ciblées des tumeurs solides, les résultats initiaux des études pivot sont trop rarement confirmés par les publications ultérieures. Ils se révèlent toujours plus favorables au nouveau médicament que les résultats finaux, témoignant que beaucoup d’essais ne représentent pas l’usage en population réelle et suggérant que certains résultats ont pu bénéficier d’un «embellissement» avant présentation aux agences de régulation.
Introduction
Depuis une quinzaine d’années, l’autorisation de mise sur le marché[4] (AMM) des thérapies innovantes (ou molécules dites ciblées) en cancérologie est prioritairement accordée selon un processus accéléré après une (ou parfois deux) études courtes dites études pivot, contre placebo sur peu de malades, jugées sur un critère de substitution[i]. Selon la doctrine « officielle », cette AMM accélérée prévoit que le laboratoire bénéficiaire présente des études complémentaires confirmant l’utilité de son médicament sous peine de perte ou de suspension de l’autorisation.
Notre étude vise à vérifier la fiabilité à long terme des résultats présentés précocément aux agences, l’efficacité du processus de contrôle et en particulier de vérifier que les études post commercialisation sont publiées, que leurs résultats sont compatibles à ceux de l’étude pivot et qu’en cas de discordance l’AMM leur est supprimée.
Méthode
Recueil des données.
Une recherche informatisée systématique a été réalisée sur Medline et Google pour identifier les études pivot portant sur une des thérapies ciblées suivantes : avastin (bevavucimab), herceptin (trastuzumab), erbitux (cetuximab), tarceva (erlotinib), nexavar (sorafenib), iressa (gefitinib,), xalcori (crizotinib), giotrif (afatinib), torisel (temsirolimus), votrient (pazopanib), sutent (sunitinib), inlyta (axitinib),(qui regroupent environ 70% des prescriptions), dans le traitement des cancers pulmonaires non à petites cellules, du rein, du colon, du sein et ORL chez l’homme (soit environ 50% des cancers non hématologiques).
Cette recherche a été inaugurée par la consultation systématique des avis des agences de régulation telles que la Food and Drug Administration (FDA), l’agence européenne du médicament European Medication Administration (EMA), le NICE institut anglais, et la Haute Autorité de Santé (HAS) pour les drogues et les cancers étudiés. La lecture des résumés obtenus a permis de répertorier les articles décrivant les essais randomisés initiaux (études pivot) et l’actualisation de leurs résultats lors d’essais dont au moins un bras comportait une des thérapies ciblées étudiées comme traitement d’un des cancers de l’étude. Leur analyse complète et la prise en compte de leurs références bibliographiques a permis d’élargir la recherche des données en particulier sur la toxicité et la qualité de vie.
Outre les descriptions des essais et de leurs résultats par les investigateurs, nous avons utilisé les macro analyses sur les drogues étudiées, et les comptes rendus de congrès de sociétés savantes, tels que ceux de l’American Association for Clinical Oncology (ASCO), l’European Society for Medical Oncology (ESMO), de la société européenne d’urologie et les articles étudiant spécifiquement la toxicité des drogues et la qualité de vie.
Nous n’avons pas recherché les essais non publiés auprès des laboratoires pharmaceutiques ou des institutions concernés, des leaders d’opinion ou des investigateurs potentiels, car nous voulions n’utiliser que des données publiques et craignions les biais liés aux conflits d’intérêts de ces sources. Tous les essais répertoriés ont ensuite été examinés pour sélectionner ceux qui satisfaisaient les critères de cette étude.
Critères de sélection.
Pour l’évaluation des résultats à long terme, n’ont été retenus que les essais de méthodologie satisfaisante, portant sur au moins 100 malades, souffrant d’un des cancers précités, vus au stade métastatique et soumis à un traitement comportant une des thérapies ciblées étudiées. La survie sans progression, la survie globale et la toxicité devaient être réévaluées lors de l’actualisation des résultats ou des études post commercialisation.
Pour évaluer le risque de biais dans les essais pris en compte, nous avons suivi les recommandations de la fondation Cochrane portant sur le tirage au sort et l’administration du traitement à l’aveugle, l’évaluation de la réponse à l’aveugle et une description complète des résultats sans sélection a postériori.
Analyse statistique.
Depuis les travaux de Buyse, les macroanalyses d’essais thérapeutiques utilisent des tests statistiques privilégiant les données numériques sur les données qualitatives. Cette approche, mathématiquement justifiée , ne nous parait pas cliniquement pertinente dans le cas précis. En effet, peu importe aux malades que le taux de réponse objective, le gain de SSP[5] ou de survie globale (OS[6]) initiaux soient statistiquement corrélés aux gains de survie ultérieurs.
Cette corrélation n’a d’intérêt pour le malade que si le gain initialement publié est garant d’un gain de survie globale pertinent en durée[7] et en qualité et d’une balance avantages/risques favorable. Pour estimer la probabilité que les discordances constatées soient uniquement dues au hasard, nous avons utilisé le log rank test .
Résultats.
La recherche bibliographique a permis de retrouver 10554 références, de lire 2300 résumés, puis 360 articles détaillant 42 études pivot. Seulement 32 d’entre elles portant sur 16872 malades remplissent les critères de sélection et constituent la base de données de cette étude.
Les raisons principales du rejet des essais publiées ont été : le caractère incomplet des données publiées et un risque de biais trop important lors de l’actualisation des résultats. Le recueil des données concernant les 32 essais retenus a nécessité de comparer l’article princeps aux publications ultérieures (au total 132 articles et rapports).
Seulement 10 des 32 études pivots présentées aux agences de régulation lors des demandes d’AMM ont vu leurs résultats confirmés totalement par les essais ultérieurs. 6 d’entre eux sont confirmés partiellement.
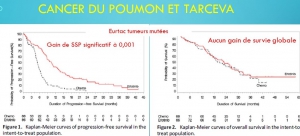
La discordance entre résultats initiaux et tardifs porte avant tout sur la toxicité. Dans la moitié des cas (16/32), la toxicité annoncée par l’étude pivot est nettement sous-estimée. Dans un tiers des cas (10/32), le gain de survie globale est également surévalué. Le bénéfice de survie sans progression(=stabilisation tumorale) est par contre généralement confirmé par les études post commercialisation.
Les discordances constatées entre résultats initiaux et tardifs se fait toujours dans le sens d’une plus grande efficacité alléguée ou d’une moindre toxicité rapportée de la nouvelle drogue dans l’étude pivot. Une telle discordance à sens unique ne peut pas être due au hasard (p< 0,0001).
Ces discordances biaisent l’évaluation de la balance avantages/risques et sont susceptibles d’expliquer en partie l’autorisation de mise sur le marché de thérapies plus toxiques qu’efficaces. Citons quelques exemples : l’avastin, l’iressa, le tarceva et l’afinitor dans le cancer du poumon, l’afinitor, le nexavar, le giotrif et le tarceva dans le cancer du sein, le styvarga, l’erbitux et le vectibix dans le cancer du colon, le votrient et le sutent dans le cancer du rein, l’erbitux dans les cancers ORL.
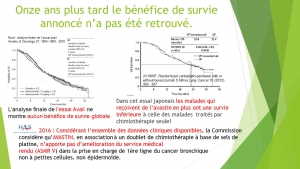
Le suivi des AMM montre enfin que très peu d’entre elles sont retirées, même lorsque l’actualisation des données montrent une balance avantages/ risque défavorable et même lorsqu’elle est actée par la Haute Autorité de Santé.
Discussion
Notre étude souffre de plusieurs limites : d’une part elle n’est pas exhaustive, elle ne porte pas sur tous les cancers ni sur toutes les thérapies ciblées. Elle n’envisage que cinq cancers et douze thérapies ciblées. Pour ces raisons, elle constitue plus un sondage qu’une macroanalyse classique.
Mais ses limites sont aussi sa force dans la mesure où elle se concentre sur la question que se pose tout clinicien : les résultats favorables d’une étude pivot constituent-ils la garantie d’une balance avantages/risques favorable pour les malades correspondant à l’indication de l’AMM ? Cette étude constate clairement que ce n’est pas le cas pour les thérapies ciblées étudiées confirmant, ainsi les travaux de Tannock[ii].
De multiples facteurs peuvent expliquer la non reproductibilité des études pivot que nous constatons dans cette étude. La sélection des malades inclus dans les essais réalisés pour obtenir l’AMM élimine les patients à risques et aboutit à un créer un échantillon peu représentatif de la population à laquelle s’adresse le médicament étudié et en particulier moins sensible aux effets secondaires.
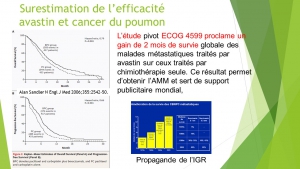
Mais il n’est pas exclu que, comme dans le cas du vioxx, certaines complications soient volontairement omises sous l’influence des sponsors ainsi que le démontre Zarin [iii] Nielsen[iv] ou Seruga[v]. Notre étude retrouve une sous-estimation de la toxicité dans 50% des études pivot. Ce chiffre, très élevé est cependant inférieur à celui de Badillo qui l’estime à 67% dans 164 essais portant sur les traitements du cancer du sein[vi].
La surestimation fréquente du gain de survie globale que nous constatons peut résulter de la méthode de recueil de l’information[vii], ou de la date choisie par les expérimentateurs pour évaluer l’effet de la nouvelle thérapie de manière flatteuse, ainsi que l’a montré Haut[viii] mais elle suggère aussi que « l’embellissement » des résultats des études pivots ne porte pas seulement sur la toxicité[ix].
Sous-estimation de la toxicité et surestimation de la survie perturbent l’appréciation de la balance avantage/risque et la complaisance des agences du médicament et en particulier de l’EMA entraîne trop souvent la mise sur le marché de drogues qui se révèlent ultérieurement inutiles et toxiques et qui continueront néanmoins à être autorisées [x].
Dans les 5 cancers étudiés, la commercialisation des douze thérapies ciblées précitées n’a apporté qu’exceptionnellement un bénéfice clinique indiscutable pour les malades. Aucun des médicaments étudiés n’est parvenu à guérir des malades métastatiques. A peine un médicament sur dix (3/32) a permis de prolonger un peu la survie de malades souffrant de tumeurs avancées et/ou d’améliorer de manière pertinente leur qualité de vie. La commercialisation trop précoce de ces drogues s’est le plus souvent soldée par une toxicité accrue sans augmentation de survie globale[xi].[xii]
l’utilisation de ces drogues en adjuvant, pour des maladies localisées opérables, s’est également révélée inefficace (à l’exception de l’herceptine), confirmant la trop faible efficacité des thérapies ciblées étudiées.
Parfois même cette mise à disposition accélérée des thérapies innovantes s’est révélée franchement délétère (12 mois de survie en moins que le placebo lors de l’utilisation de l’iressa en consolidation[xiii], 12,7 mois de survie en moins pour les patients de l’essai luxlung 3 porteurs de la mutation EGFR L858R traités par giotrif (27.6 mois vs.40.3 mois), 3 mois de survie en moins que la chimiothérapie avec le tarceva dans les cancers du poumon sans sélection génétique[xiv], 45 jours de survie en moins et une dégradation de la qualité de vie lors de l’addition d’erbitux à la chimiothérapie des cancers du colon[xv]) du fait d’indications inadaptées qui auraient pu être évitées si une plus longue période d’essai avait permis de mieux préciser les indications possibles.
Une amélioration du processus d’AMM est nécessaire La commercialisation accélérée, sur des critères substitutifs peu représentatifs de la survie globale nuit plus souvent aux malades qu’elle ne leur est utile. Les critères d’acceptation des nouvelles drogues doivent être relevés pour mieux protéger la population[xvi] [xvii].
En particulier, les critères d’inclusion des patients dans les études pivot doivent permettent de réaliser des groupes de malades représentatifs de la population à laquelle s’adresse le médicament. Une attention accrue des agences est nécessaire pour s’assurer que les toxicités soient totalement rapportées. Les évaluations intérimaires et les publications trop précoces de données immatures doivent être limitées et ne plus être acceptées pour la délivrance d’AMM. Il parait enfin indispensable que des études post commercialisation soient réalisées par des organismes indépendants des firmes, afin de vérifier la véracité des résultats allégués lors des études pivot et de corriger éventuellement les AMM.
Conclusions.
Lors d’essais pivot (essais cliniques) de thérapies ciblées (molécules innovantes) en cancérologie, les résultats sont biaisés par la sélection des malades inclus. Leur présentation aux agences minore habituellement la toxicité et surestime fréquemment l’efficacité faussant ainsi l’évaluation de la balance avantages/ risques. L’AMM ne constitue donc plus un gage certain d’utilité pour les malades, et il est souvent plus pertinent d’utiliser préférentiellement des médicaments moins récents, pour lesquels on dispose d’informations confirmées.
[1] « étude clinique (ou un essai clinique) : situation expérimentale au cours de laquelle on teste chez l’homme la véracité ou non d’une hypothèse. Essai clinique sur un médicament vise à mettre en évidence ou à en vérifier les effets et (ou) à identifier tout effet indésirable et (ou) à en étudier l’absorption, la distribution, le métabolisme et l’excrétion pour en définir l’efficacité et la sécurité d’emploi » : définition du LEEM http://www.leem.org/sites/default/files/EtUDES%20CLINIQUES20questionsmai200%5B1%5D.pdf. Malheureusement ces nouvelles études sont très rapides sur quelques mois, et ne permettent pas le recul suffisant pour dépister les effets secondaires tardifs.
[2] https://docteur.nicoledelepine.fr/les-resultats-des-etudes-pivot-de-therapies-ciblees-en-cancerologie-des-tumeurs-solides-sont-ils-fiables-2/ Présentation au colloque de Bobigny sur surmédicalisation en avril 2017 diaporama disponible à l’adresse à copie-collé
[3] G. Delépine N. Delépine , S Alkhallaf
[4] AMM
[5] Survie sans progression, nouvelle formulation de maladie stable ou SD stable disease.
[6] OS = overall survival ou survie globale
[7] Et non seulement de quelques jours comme trop souvent le cas.
Références médicales
[i] Biologics Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) May 2007
[ii] Tannock Relevance of randomised controlled trials in oncology. Lancet Oncol. 2016 Dec;17(12)
[iii] Zarin DA, Tse T. The effect of funding source on outcome reporting among drug trials. Ann Intern Med 2011;154(2):137-8
[iv] Als-Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomised drug trials. A reflection of treatment effect or adverse events? JAMA 2003; 290: 921-28.
[v] Seruga.B Under-reporting of harm in clinical trials. Lancet Oncol. 2016 May;17(5)
[vi] F. E. Vera-Badillo, R. Shapiro, A. Ocana, E. Amir & I. F. Tannock Bias in reporting of end points of efficacy and toxicity in randomized, clinical trials for women with breast cancer Annals of Oncology 24: 1238–1244, 2013
[vii] Doria-Rose VP, Marcus PM, Miller AB, Bergstralh EJ, Mandel JS, Tockman MS, et al. Does the source of death information affect cancer screening efficacy results ? A study of the use of mortality review versus death certificates in four randomized trials. Clinical Trials 2010;7(1):69-77
[viii] Haut ER, Pronovost PJ. Surveillance bias in outcomes reporting. JAMA 2011;305(23):2462-3.
[ix] Berger E. Ghostwriters, data manipulation and dollar diplomacy: how drug companies pull the strings in clinical research. Ann Emerg Med 2008;52(2):137-9
[x] Banzi R, Gerardi C, Bertele V, Garattini S Approval of drugs with uncertain benefit risk profiles in Europe”, Eur J Intern Med, 2015;26:572-84.
[xi] Light DW, Lexchin J. Why do cancer drugs get such an easy ride?
BMJ 2015;350:h2068
[xii] Apolone G, Tafuri G, Trotta F, et al. A new anti-cancer drug in the
market: good news for investors or for patients? Eur J Cancer
2008;44:1786–8.
[xiii] Karen Kelly, Kari Chansky, Laurie E. Gaspar, Kathy S. Albain, James Jett, Yee C. Ung, Derick H.M. Lau, John J. Crowley, and David R. Gandara Phase III Trial of Maintenance Gefitinib or Placebo After Concurrent Chemoradiotherapy and Docetaxel Consolidation in Inoperable Stage III Non–Small-Cell Lung Cancer: SWOG S0023 J Clin Oncol 26:2450-2456. © 2008
[xiv].Garassino MC1, Martelli O, Broggini M, Farina G, Veronese S, Rulli E, Bianchi F, Bettini A, Longo F, Moscetti L, Tomirotti M, Marabese M, Ganzinelli M, Lauricella C, Labianca R, Floriani I, Giaccone G, Torri V, Scanni A, Marsoni S; TAILOR trialists.Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. Lancet Oncol. 2013 Sep;14(10):981-8
[xv] Colien Tol, M.D., Miriam Koopman, M.D., Annemieke Cats, et al Chemotherapy, Bevacizumab, and Cetuximab in Metastatic Colorectal Cancer N Engl J Med 2009;360:563-72.
[xvi]BEUC A FAST-TRACK APPROVAL FOR NEW MEDICINES – PATIENT SAFETY AT RISK? BEUC position on adaptive pathways Ref: BEUC-X-2016-066 – 01/07/2016
[xvii] “A C Davis, J Lexchin, T Jefferson, P Gøtzsche, M McKee Adaptive « pathways” to drug authorisation: adapting to industry? BMJ 2016;354:

olloque bobigny 2017 resultats-pivots