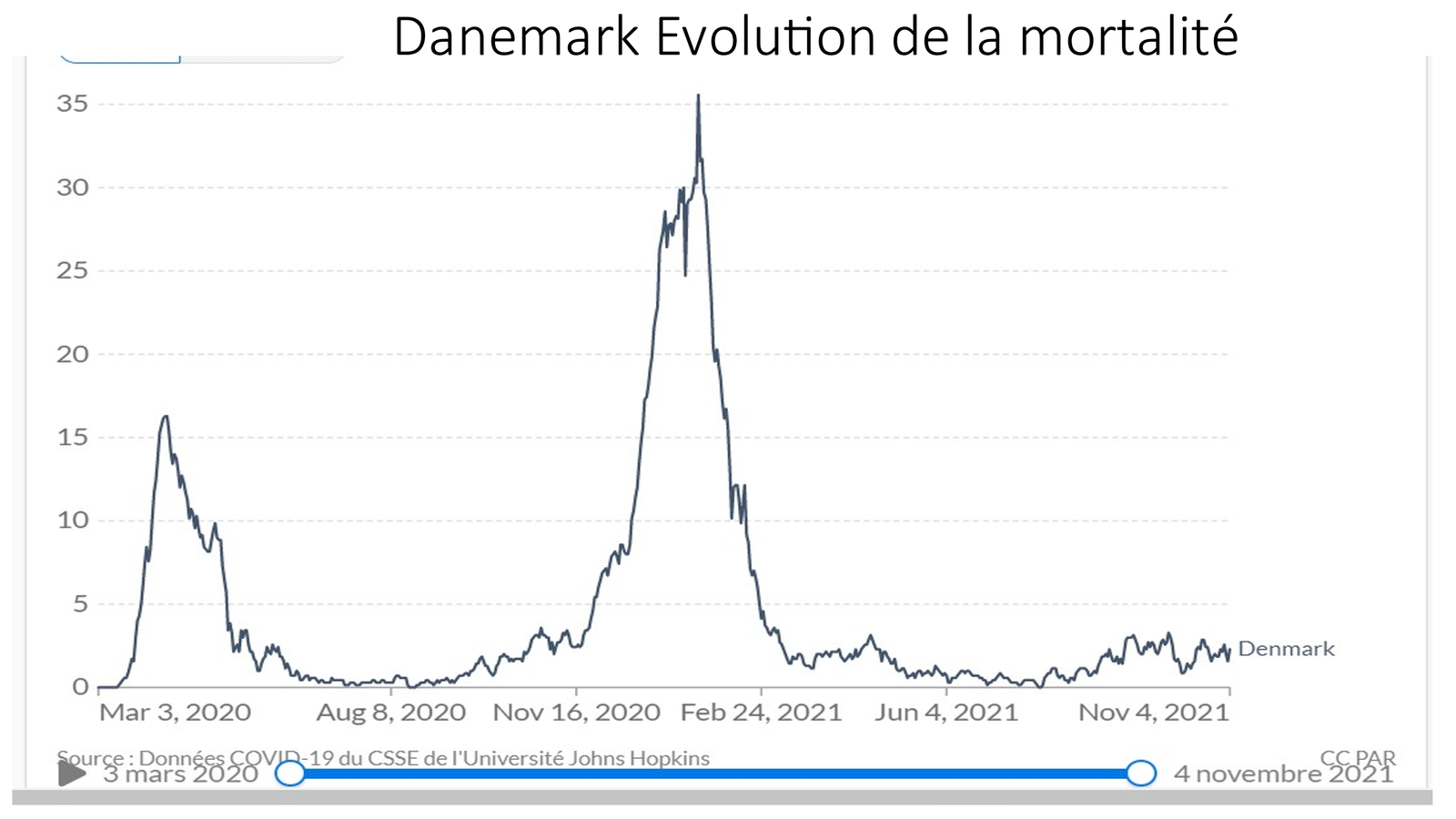
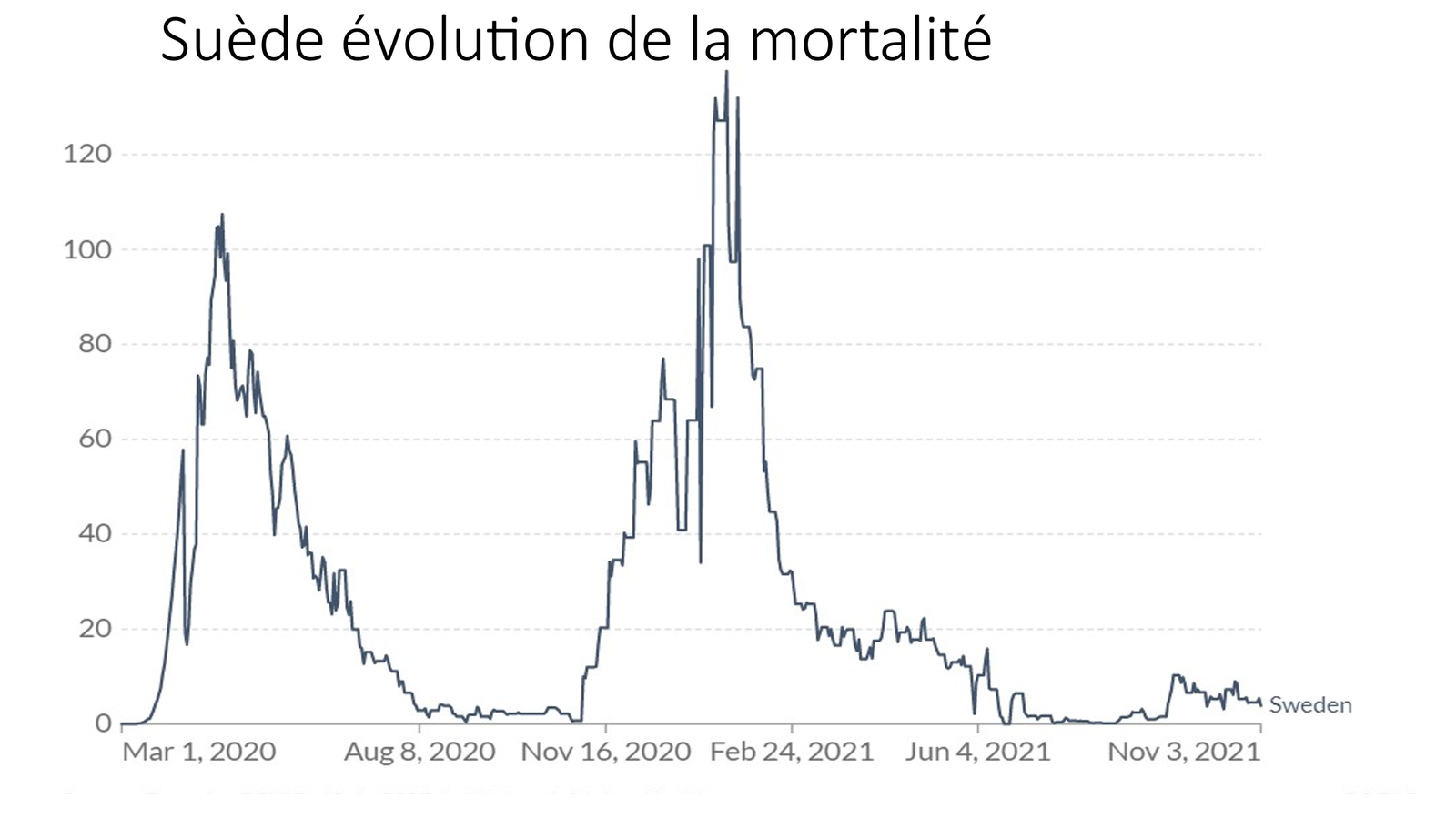
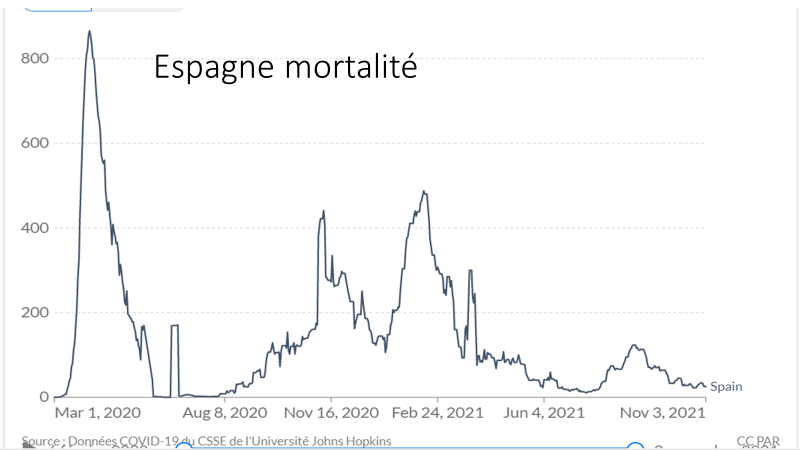
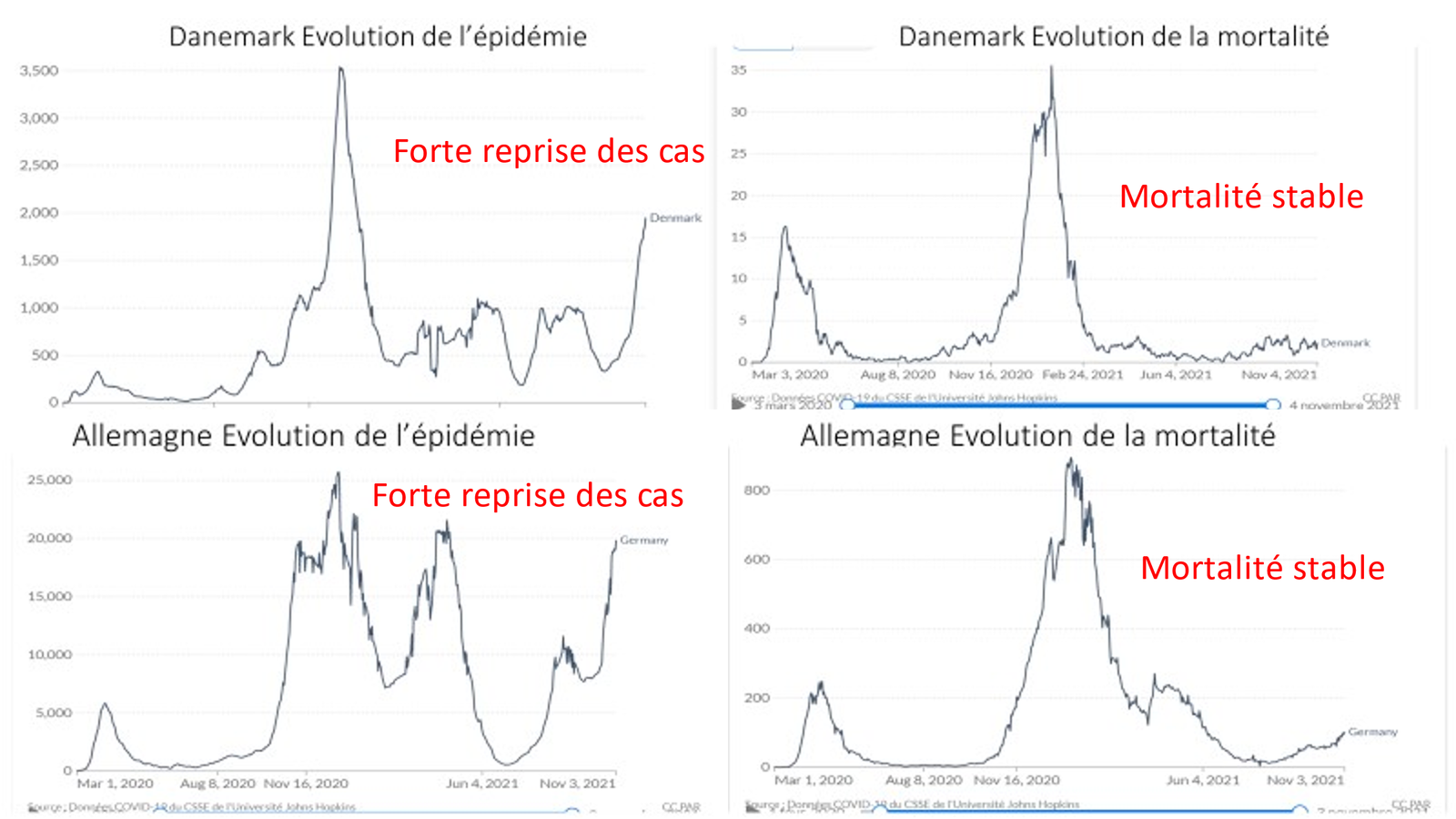
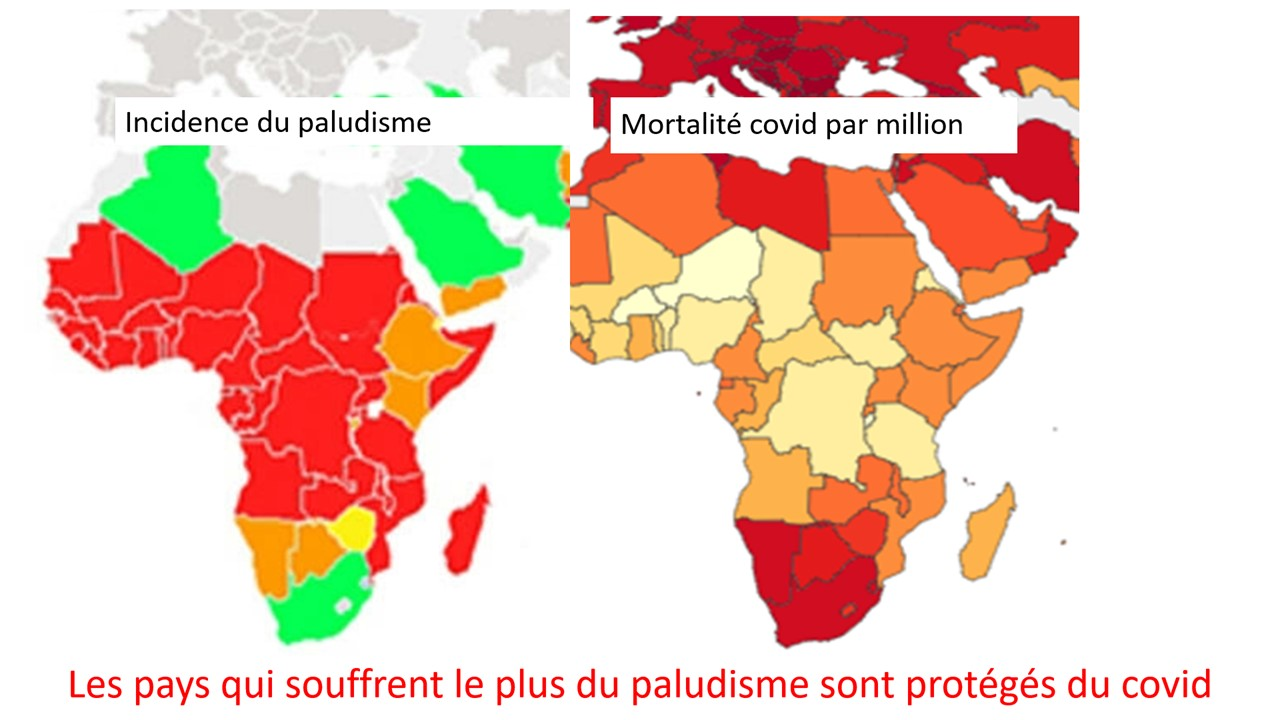
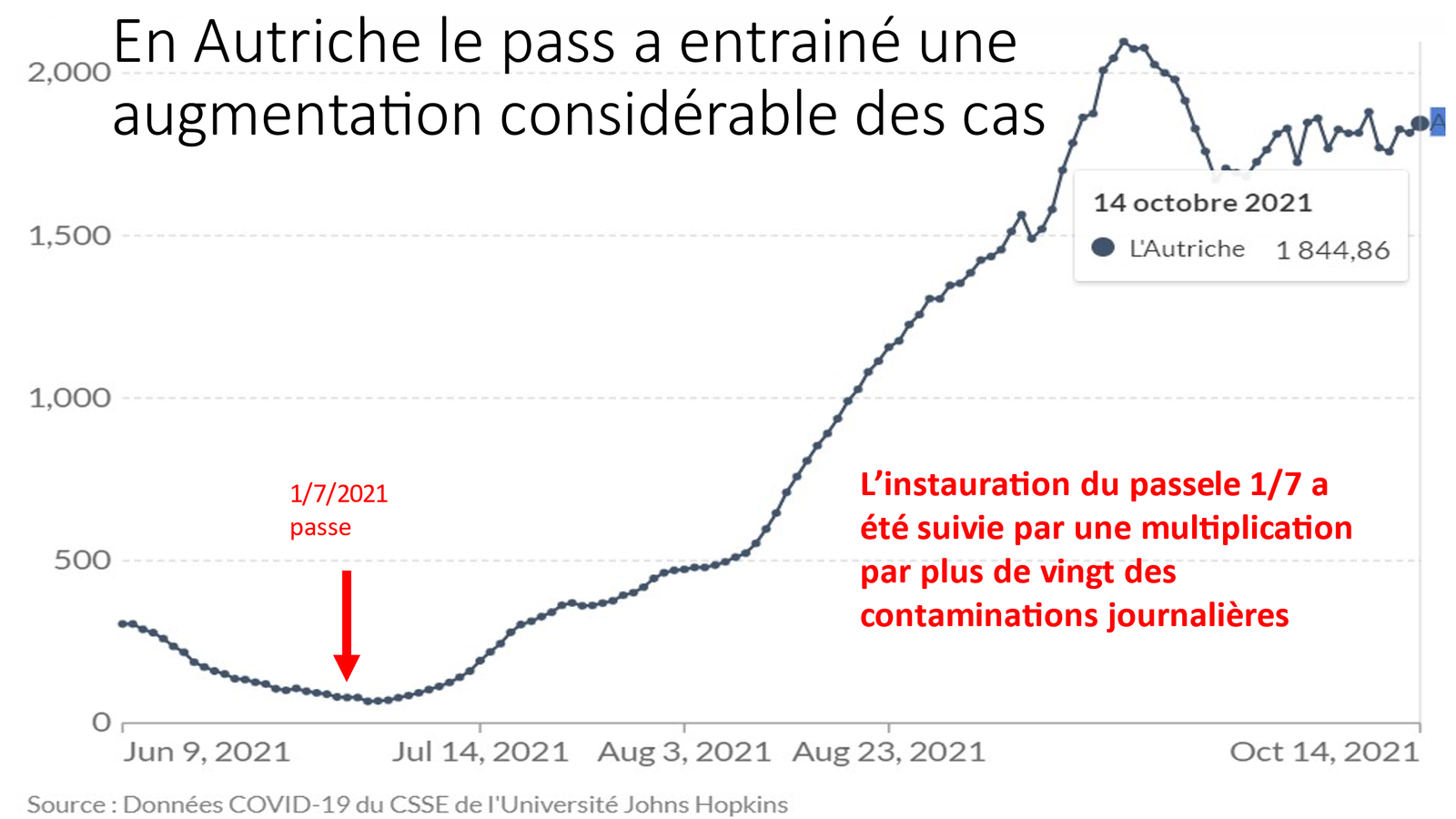
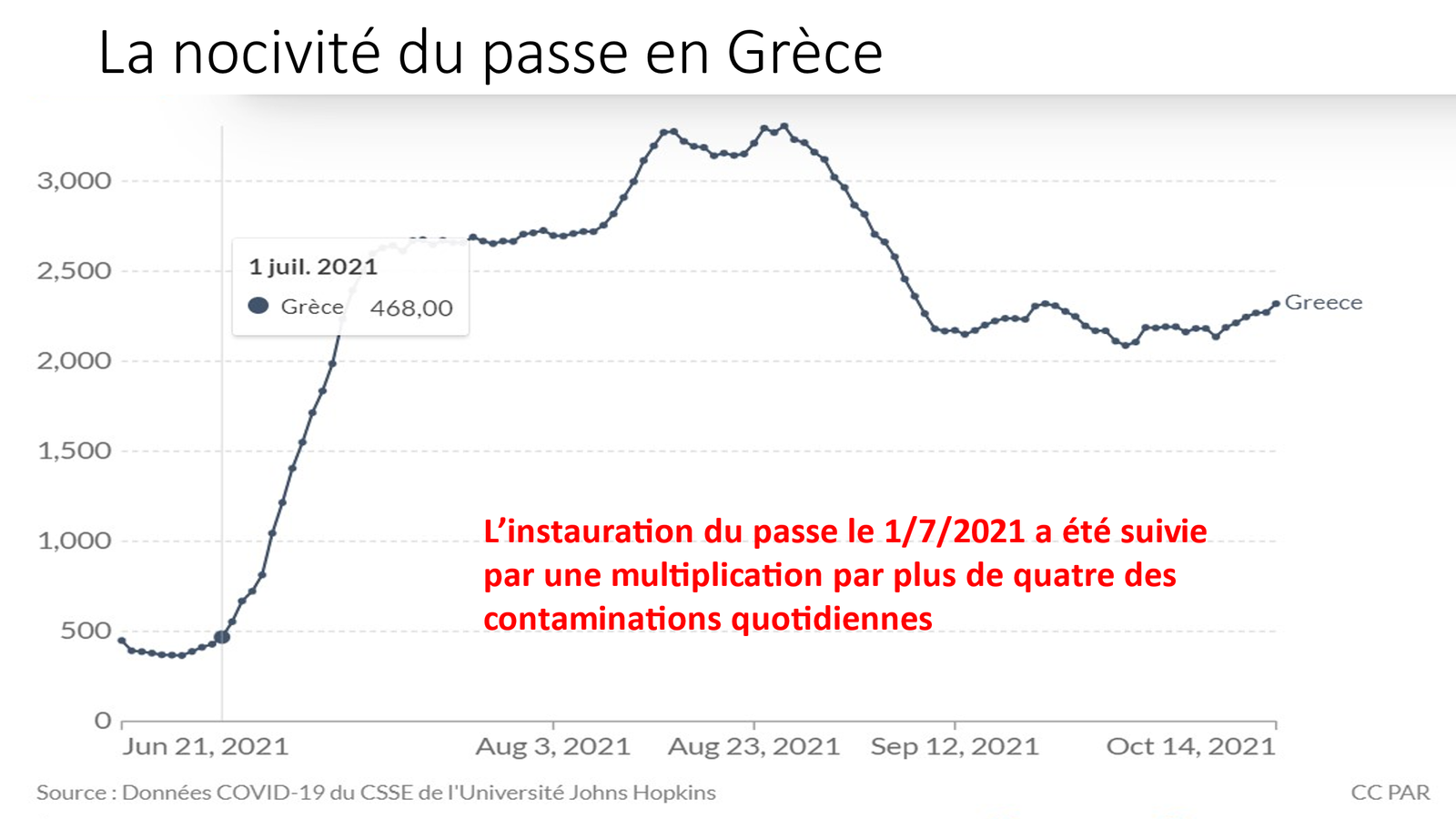
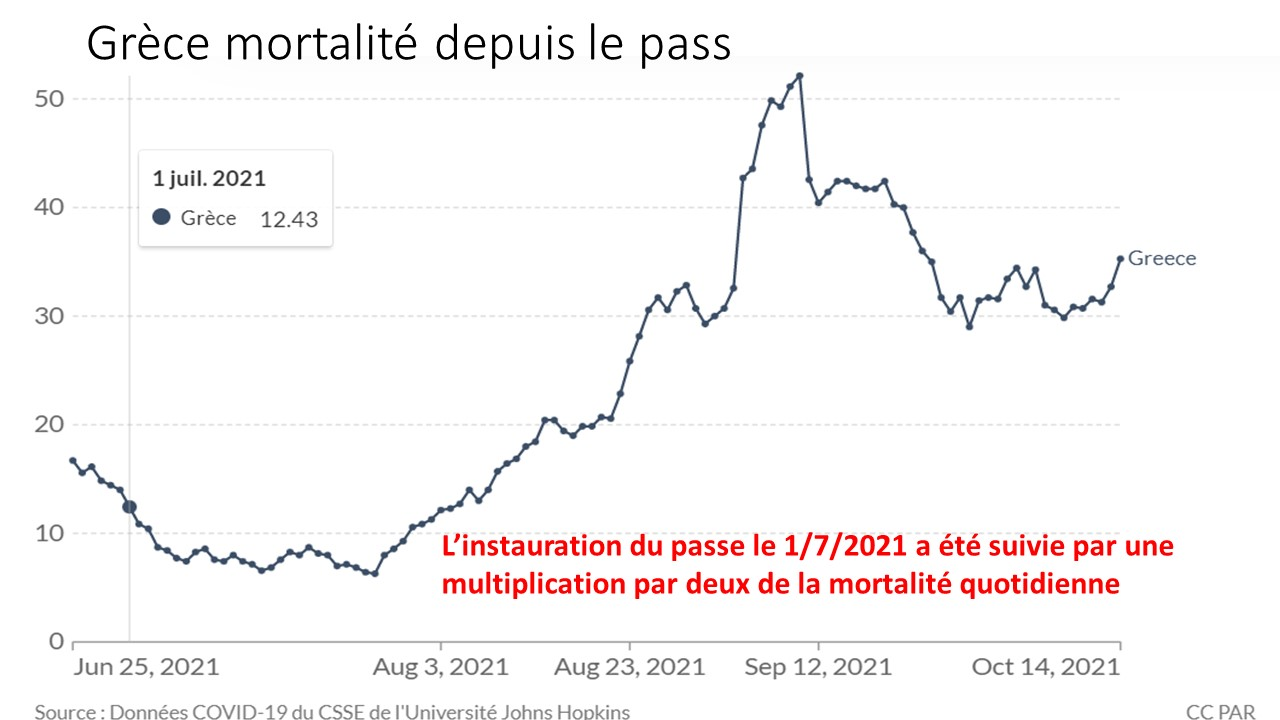
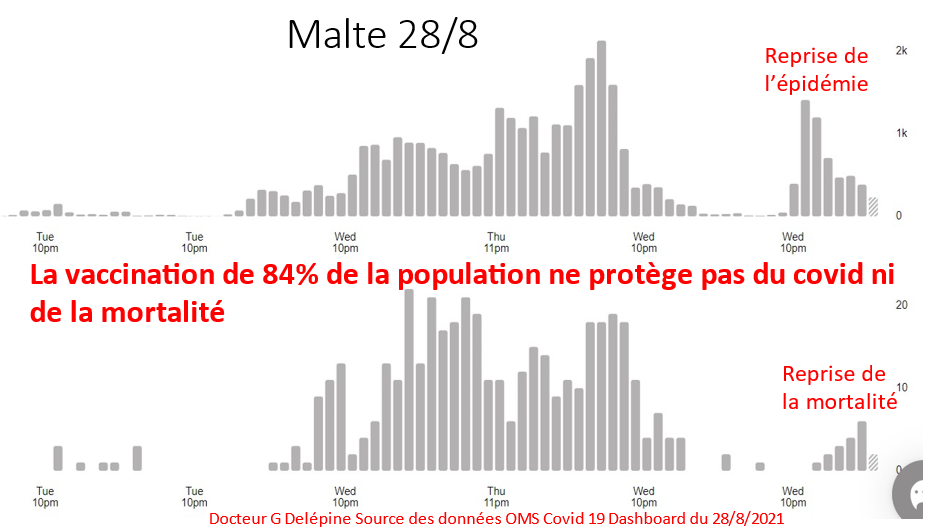
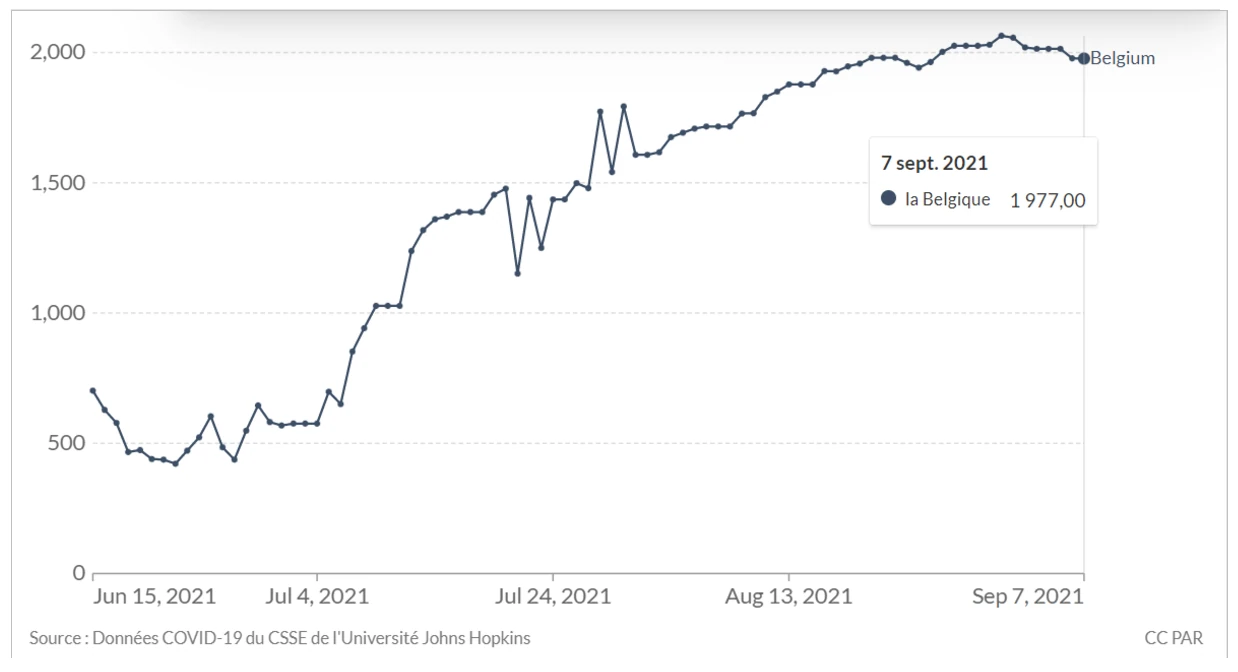
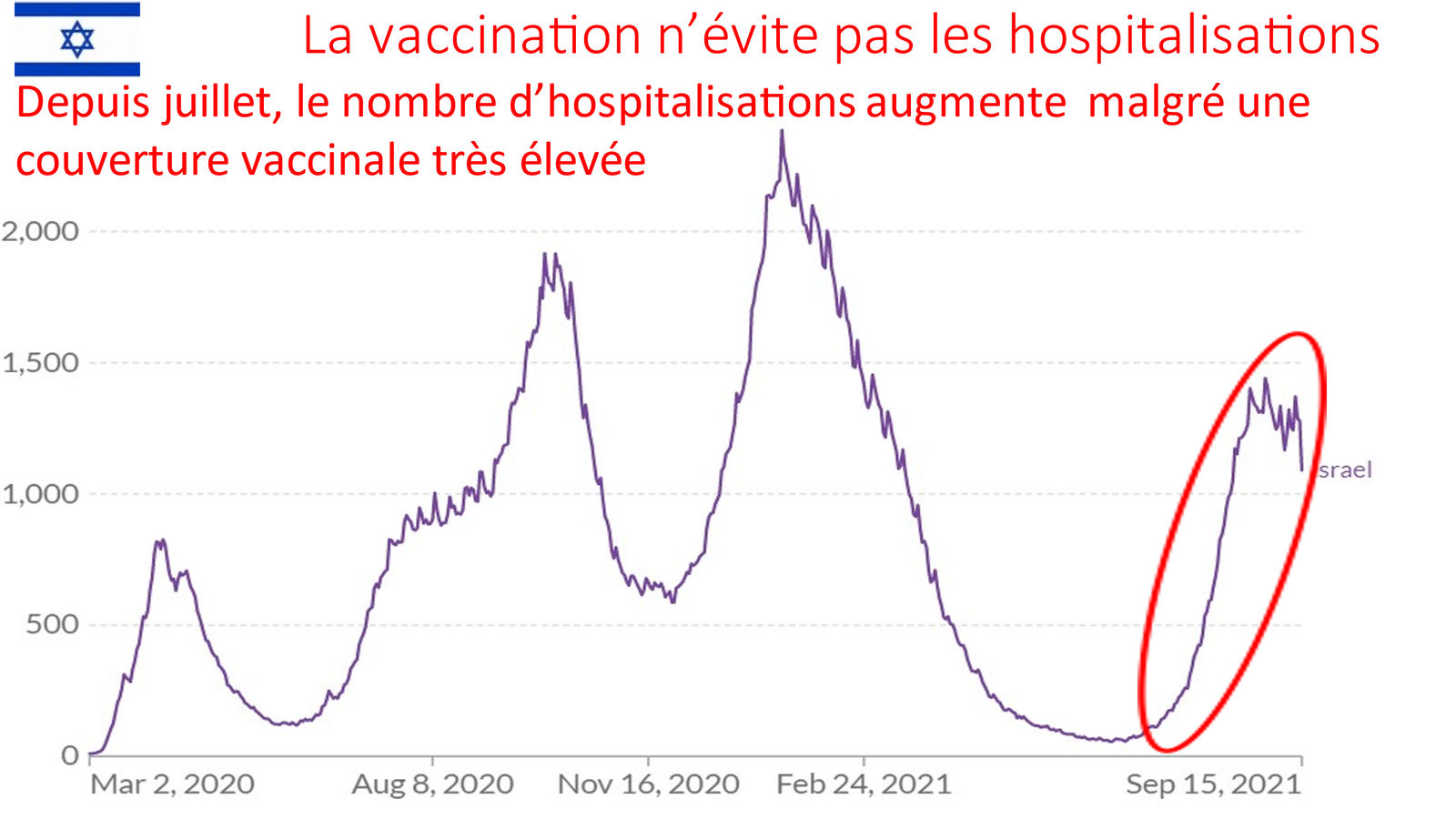
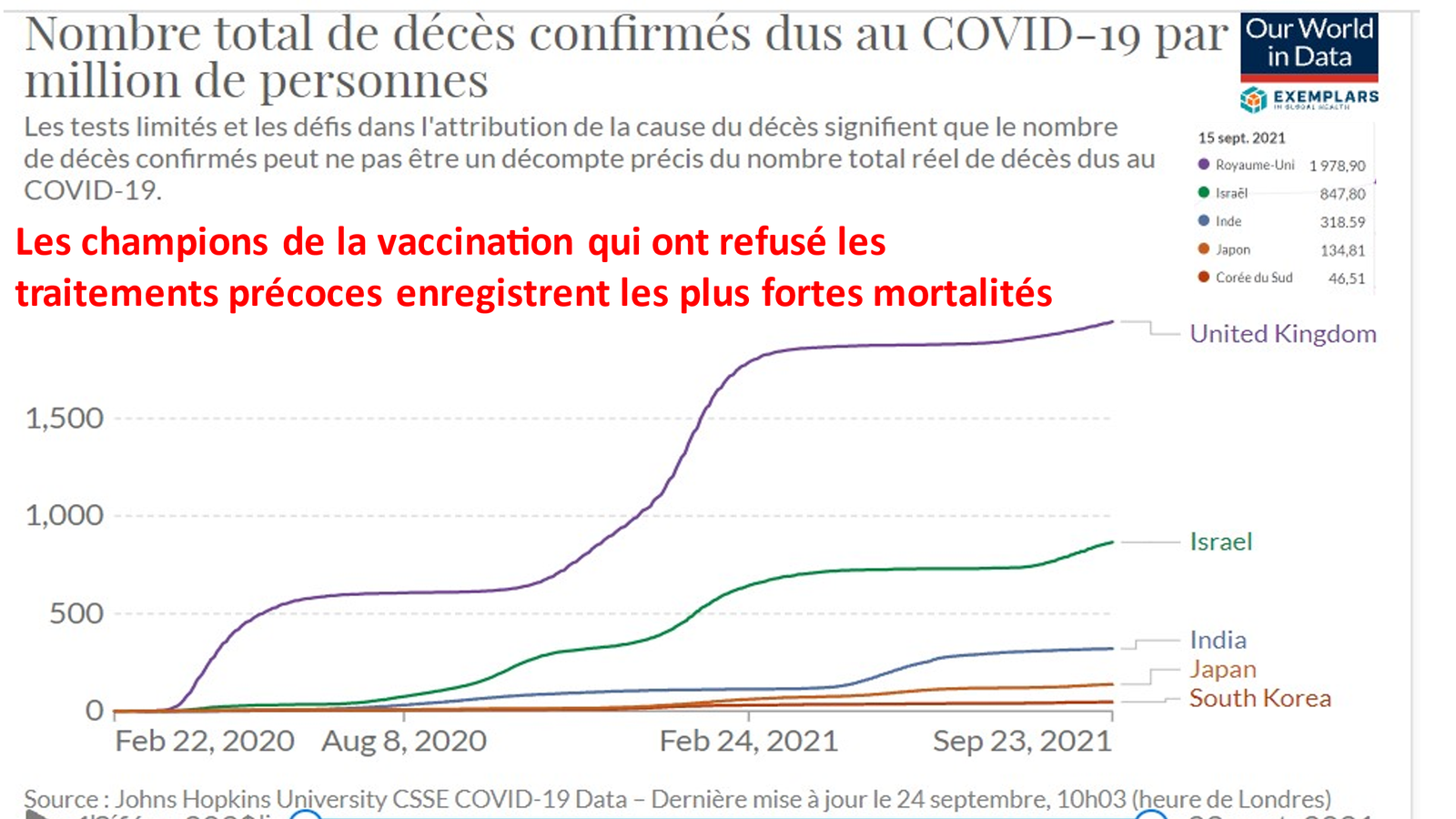
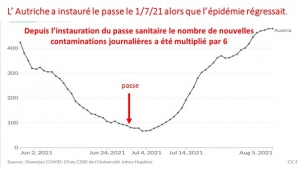
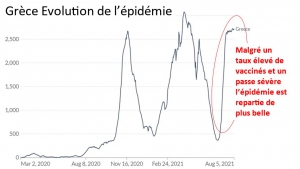
Laisserons-nous les enfants courir le risque d’atteinte cardiaque post pseudovaccin Covid pour sauver une sortie au resto ? EPEE DE DAMOCLES SUR NOS ENFANTS
Ou même une inscription dans un club de foot ?
Laissez-les jouer devant l’immeuble avec les copains ou courir les champs si vous êtes à la campagne. Rien ne vaut la santé, et on a vécu sans salle de gym avec équipements lourds pour COURIR, sans club payant !
Un adulte sympathique aidait les gosses dehors à faire des passes … La coronafolie va bien se terminer et d’autant plus vite que nous ne céderons pas.
Ne risquez pas l’avenir de votre petit, pour quelques mois de vie « différente » et pas « anormale », car finalement courir sur un tapis fixe est -il plus normal qu’une balade dans les bois ? Ce sera l’occasion de leur expliquer la vie d’autrefois, et la gabegie actuelle plutôt que des grands cinémas pro-planète… Les bobos parisiens ont-ils renoncé à leur SUV pour se promener et à leurs clubs de sport consommateurs d’énergie ? En tous cas les milliardaires n’ont pas renoncé à leurs jets privés pour se montrer et faire croire qu’ils veulent sauver la planète, mais c’est un autre sujet (apparemment).
DES ALERTES MULTIPLES (sauf en France, comme pour Tchernobyl ?)
Info : une nouvelle catastrophe
F-Dagoury, meilleur plongeur en apnée du monde, a été diagnostiqué d’une myocardite et d’une péricardite 40 jours après sa deuxième dose Pfizer. Le plongeur a consulté un cardiologue qui lui a dit que c’était un effet secondaire courant du Pfizer[1]
L’agence de presse allemande dresse une liste de 75 athlètes européens décédés « soudainement » au cours des 5 derniers mois depuis qu’ils ont été complètement vaccinés.[2]
« Nous avons pris beaucoup de temps pour cette recherche, en choisissant chaque cas individuellement. Y a-t-il eu un nombre évident de décès « soudains et inattendus » dans le sport et le sport d’élite seulement à la mi-2021 ? Une accumulation possible depuis le début des vaccinations géniques peut (et ne veut pas) être expliquée par n’importe qui du côté officiel. Selon la lecture des médias et de la politique, peut-être victimes de chantage et de pots-de-vin, ce sont des coïncidences malheureuses – bien que même les jeunes de 13 ans souffrant de problèmes cardiaques tombent sur le terrain. »
L’agence de presse poursuit, par objectivité :
« Nous voudrions commencer ce rapport par les arguments de l’autre partie, car il est toujours important d’écouter toutes les parties. Nous voudrions également recommander ce principe à la presse judiciaire et au système, même s’il n’y a pas d’importantes subventions d’État à utiliser pour cela.
En 2016, le média français Futura Santé s’est plaint de « nombreux » décès cardiaques dans le football. Elle toucherait 2 athlètes sur 100 000 par an en France, soit un total de 1 000 à 1 500 personnes. Le « New Statesman » parle de 12 jeunes qui meurent de mort subite d’origine cardiaque en courant – chaque semaine. L’article a été publié en novembre 2018 en Angleterre. En février 2019, le journal autrichien der Standard a demandé pourquoi la mort cardiaque survient « si fréquemment » dans le sport.
« ll n’existe pas de statistiques fiables à des fins de comparaison. Mais dans Wikipédia, il y a des listes d’athlètes qui sont morts pendant le jeu. Cette liste remonte à 1889 et est longue à première vue – mais en fin de compte, il s’avère que même dans les « années de catastrophe », environ 5 à 8 décès de ce type ont été enregistrés ».
Y a-t-il plus de gens qui meurent actuellement « soudainement et de manière inattendue » dans le sport, surtout lorsqu’il s’agit de professionnels et d’athlètes de haut niveau ? Et est-ce à cause de la vaccination ? ou bien avons-nous cauchemardé ?
Plus de 75 cas connus au cours des 5 derniers mois[3]
- 4.6.21, Italie: L’ex-professionnel Giuseppe Perrino, 29 ans, s’effondre lors d’un match de charité pour son frère décédé et meurt.
- 7.6.21, Allemagne 38 ans Le professionnel de tennis de table Michael Schneider meurt subitement et de manière inattendue.
- 12.6.21, Danemark, 29 ans Le footballeur Christian Eriksen s’effondre sans vie lors d’un match de championnat d’Europe – il peut être relancé, a besoin d’un stimulateur cardiaque pour le reste de sa vie.
- 22.6.21, Hongrie, Le footballeur de 18 ans ViktorMarcell Hegedüs est décédé alors qu’il s’échauffait pour un entraînement en Hongrie.
- 14.07.21, Pays-Bas, 31 ans champion olympique de patinage de vitesse Kjelt Nuis gravement malade après la vaccination,avec des problèmes cardiaques à l’hôpital.
- 16.07.21, Le footballeur égyptien Imad Bayumi est décédé lors d’un match amical en Égypte.
- 22.07.21, Allemagne, 36 ans le SV Olympia Schlanstedt et Germania de Kroppenstedt se sont rencontrés. Pendant le match, le joueur de Schlanstedt, Nicky Dalibor, s’est effondré et a dû être réanimé sur le terrain.
- 23.07.21, Allemagne, 27 ans TimB. du SV Hamberge (Schleswig-Holstein) s’effondre après son retour d’un tournoi de football et meurt.
- 24.07.21,Allemagne Un joueur du TuS Hoberge-Uerentrup (Bielefeld) s’effondre sur le terrain avec un arrêt cardiaque.
- 31.07.21, Pays-Bas, La handballeuse whitnée Abriska, âgée de 19 ans, est décédée d’un arrêt cardiaque juste avant un vol.
- 02.08.21, Belgique, 18 ans Rune Coghe de Eendracht Hoglede (Belgique) souffre d’une crise cardiaque pendant le match
- 02.08.21, Autriche, 18 ans Un joueur anonyme de 18 ans dans le Burgenland (Autriche) s’effondre sur le terrain de jeu et peut être sauvé grâce à l’utilisation d’un hélicoptère.
- 06.08.21, Allemagne Kreisliga joueur de SpVgg. Oelde II doit être relancé par son adversaire.
- 14.08.21, Belgique,37 ans, l’ancien footballeur professionnel français Franck Berrier est décédé de plusieurs crises cardiaques alors qu’il jouait au tennis.
Etc.. Suite sur l’article de référence :
https://report24.news/ab-13-jahren-lange-liste-ploetzlich-verstorbener-oder-schwerkranker-
Peut-on vraiment croire au hasard et discuter doctement de l’imputabilité alors que des familles sont endeuillées et des enfants sacrifiés au nom de la soumission aux directives dites sanitaires. Jusqu’à quel degré l’aveuglement aide-t-il à vivre ?
Les atteintes cardiaques chez l’enfant sont rares
Contrairement à ce qu’essaient de vous faire croire les médias, les atteintes cardiaques du cœur survenant brutalement chez l’enfant en bonne santé étaient devenues exceptionnelles depuis la disparition du rhumatisme articulaire aigu (les années 60) grâce au traitement par pénicilline des angines à streptocoque, en l’absence de comorbidité comme des maladies auto-immunes ou des lésions post médicamenteuses (chimiothérapie anticancéreuse par exemple).
L’incidence des myocardites chez l’enfant, estimée entre 2007-2016 aux USA est de 1/100 000.[4]
Même les maladies cardiaques congénitales s’étaient raréfiées dans nos contrées via le dépistage anténatal (et beaucoup d’avortements à la moindre communication interauriculaire) et via les interventions précoces pour les enfants nés avec des anomalies acceptées par la famille. Bref le concept de « maladie bleue » par exemple ne vous était plus familier et tant mieux.
Actuellement, les myocardites et péricardites peuvent se rencontrer dans le cadre de maladies virales, mais sont alors le plus souvent bénignes et de courte durée et plus fréquemment chez des sujets fragiles, voire immunodéprimés.
Plus exceptionnellement se rencontre en pédiatrie une myocardite grave et menaçante à l’occasion d’un diagnostic difficile qui débouche sur une maladie congénitale exceptionnelle.
Nous sommes alors dans le cadre de maladies rares, dites orphelines[5] et non de pathologies habituelles comme on aimerait vous le faire croire pour minimiser la tragédie actuelle, conséquence de pseudo vaccination inutile et dangereuse.
Donc une nouvelle fois, le récit médiatique démarre par d’énormes mensonges.
Non, les crises cardiaques qui tombent du ciel ne sont pas habituelles chez les jeunes qui jouent au foot dans la cour.
Vous avez tous des enfants à l’école ou des petits-enfants. Combien de fois par le passé récent (avant la mascarade Covid), avez-vous entendu parler de décès brutal au cours d’un cours d’éducation physique selon le vieux terme ? Jamais ! Revenez sur terre et cessez de croire aux balivernes tragiques des journalistes corrompus ou devenus complètement fous via la coronafolie.
Qu’est-ce au fond qu’une myocardite ?
Les myocardites et les péricardites sont des inflammations du muscle cardiaque (myocardites) ou de son enveloppe, le péricarde (péricardites). Classiquement elles se révèlent par une fatigue croissante, des douleurs thoraciques, des difficultés de plus en plus importantes à respirer, une accélération du rythme cardiaque, une baisse de la pression artérielle, et parfois des douleurs dans les membres. La maladie est parfois accompagnée de fièvre, car une infection virale ou bactérienne peut être à l’origine de l’atteinte cardiaque. Ce n’est pas le tableau le plus courant actuellement.
Elles peuvent passer inaperçues et ne se révéler brutalement que par un énorme malaise, voire une mort subite, à l’occasion d’un effort. Ce sont les jeunes sportifs qui paient actuellement un lourd tribut. Et les médecins belges recommandent depuis plusieurs mois de ne pas faire d’efforts au moins dans la semaine suivant l’injection.
L’évolution de la myocardite peut être très variable. Elle ne provoque souvent que des symptômes mineurs, mais elle peut aussi entraîner un arrêt cardiaque brutal ou guérir en laissant des séquelles permanentes responsable dans 20% des cas de mort dans les 5 ans qui suivent pour les myocardites habituelles.
Il est évident que les notions générales évolutives citées ici sont liées aux myocardites infectieuses ou congénitales connues et que personne ne peut prédire les conséquences de l’atteinte cardiaque par la protéine spike créée via l’injection génique nouvelle. Combien de temps le vacciné produit-il de protéines spike susceptibles d’aggraver la maladie et/ou de se compléter par d’autres lésions sur d’autres organes ? Aucune réponse claire possible sans recul.
Les prédictions de guérison possibles basées sur l’évolution des myocardites d’autres origines n’ont aucune valeur. Il serait honnête d’avouer que l’on ne sait pas ! Ce qui est certain est qu’un énorme doute existe sur l’avenir de ces patients.
ATTENTION AU BÉNÉFICE/RISQUE
Les marchands de vaccin aimeraient nous faire croire et l’écrivent sans remords, que le Covid donne plus de risque de myocardite que le vax. Ils inventent et/ou manipulent les données, oubliant que les jeunes ne meurent pas de Covid, alors que les jeunes vaccinés meurent de crise cardiaque. Regardons les données.
Quels sont les risques de myocardite après infection Covid chez les jeunes
La myocardite (ou péricardite ou myopéricardite) résultant d’une infection primaire à la Covid19 s’est produite à un taux de 450 par million chez les jeunes hommes malades du Covid. Les jeunes hommes infectés par le virus sont plus susceptibles de développer une myocardite que ceux qui ont reçu le vaccin nous dit-on.[6]
Mais ce pourcentage omet de préciser que la maladie clinique du Covid est exceptionnelle chez le jeune de moins de 20 ans et que ces atteintes cardiaques sont bénignes et d’évolution favorable rapide.
Il serait honnête d’avouer que le Covid chez le jeune ne tue pas alors que la myocardite post-vaccinale d’un sujet sain non malade tue parfois et confisque l’avenir du jeune en tous les cas. Comparaison purement chiffrée n’est pas raison.
La myocardite du vaccin antiCovid
L’histoire médiatique de l’atteinte cardiaque post vaccination remonte à quelques mois après le début des injections intensives vers mai 2021.
Fin mai 2021, le comité de sécurité des vaccins de l’Organisation mondiale de la santé (OMS) a publié une déclaration d’intérêt concernant les cas signalés d’inflammation myocardique et/ou péricardique survenant chez des patients après vaccination avec des vaccins à ARNm Covid-19. Cela faisait suite à un petit nombre de cas signalés en Israël et aux États-Unis.
Les analyses aux USA ont confirmé que le nombre de cas observés dépassait le nombre attendu de cas dans le groupe d’âge des 16-24 ans. Les cas semblaient être principalement chez les hommes et se développer peu de temps après l’administration de la 2e dose.
Démonstration de l’imputabilité de l’atteinte cardiaque à la vaccination
Les Canadiens ont vérifié chez l’adulte, sur deux mois de juin à fin juillet 2021 la survenue de myocardites après injection, ce qui a permis d’établir clairement[7] la temporalité entre l’apparition de la myocardite et la vaccination.
Les symptômes constatés chez les cardiaques canadiens sont résumés dans le tableau tiré de leur article.
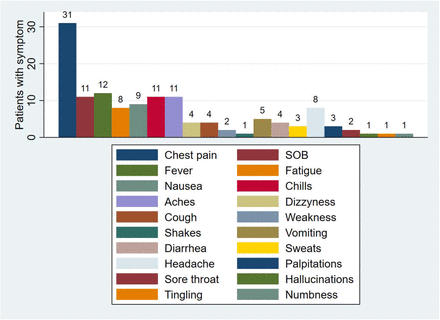
Il s’agit de la plus grande série de la littérature à établir clairement la relation temporelle entre la vaccination par l’ARNm Covid, les symptômes cliniques de la myocardite et les résultats de l’IRM.
Chez la plupart des patients, l’apparition des symptômes a commencé dans les premiers jours suivants la vaccination avec des anomalies correspondantes des biomarqueurs et de l’ECG. L’IRM cardiaque a confirmé des modifications myocardiques et péricardiques aiguës avec la présence d’un œdème démontré à la fois par la cartographie des tissus et le rehaussement tardif du gadolinium. Les symptômes se sont résorbés rapidement avec le traitement standard et les patients ont pu sortir en quelques jours.
Si la cohorte canadienne avait capturé tous les cas dans la région d’Ottawa, l’incidence de la myocardite serait de 0,1 % de toutes les doses de vaccin (32 cas/32 379 doses x 100), ou 10 cas de myocardite pour 10 000 doses de vaccin. Cette étude concerne des adultes, mais est intéressante pour notre sujet, car elle confirme le lien entre atteinte cardiaque et injection génique.
L’une des cohortes les plus importantes à ce jour a décrit la nécessité d’un traitement de l’insuffisance cardiaque chez 40 % de ses patients atteints de myocardite (malgré l’absence d’épisodes d’insuffisance cardiaque antérieurs) et de soins intensifs chez 10 %. Mais l’étude concerne les adultes et la maladie cardiaque post vaccinale paraît d’autant plus grave que le sujet est plus jeune.
L’inflammation péricardique est un épiphénomène d’inflammation myocardique ; cela a également été démontré dans la série de cas de Starekova et al. La véritable péricardite diffuse isolée est rare dans la littérature.
La myocardite a été décrite comme complication de vaccins classiques, mais dans des proportions beaucoup plus faibles qu’avec les pseudovax antiCovid
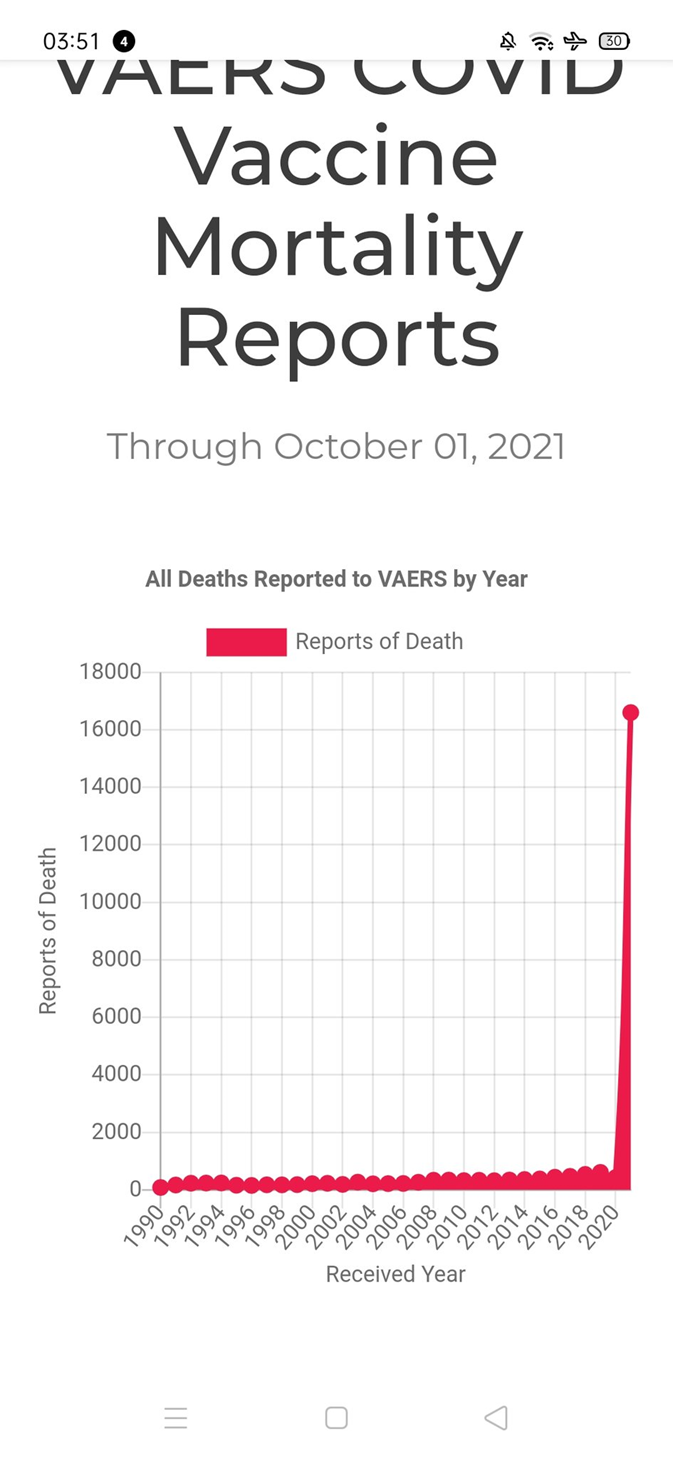
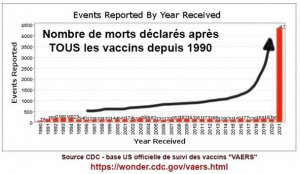
La myopéricardite est une complication reconnue associée à quelques vaccins, aux États-Unis représentant 0,1% de tous les effets indésirables collectés par le système de notification des effets indésirables des vaccins (VAERS).[8] Ces rapports, recueillis entre 1990 et 2018, révèlent que 79 % des cas concernaient des hommes et sont survenus principalement dans les 2 semaines suivant la vaccination.
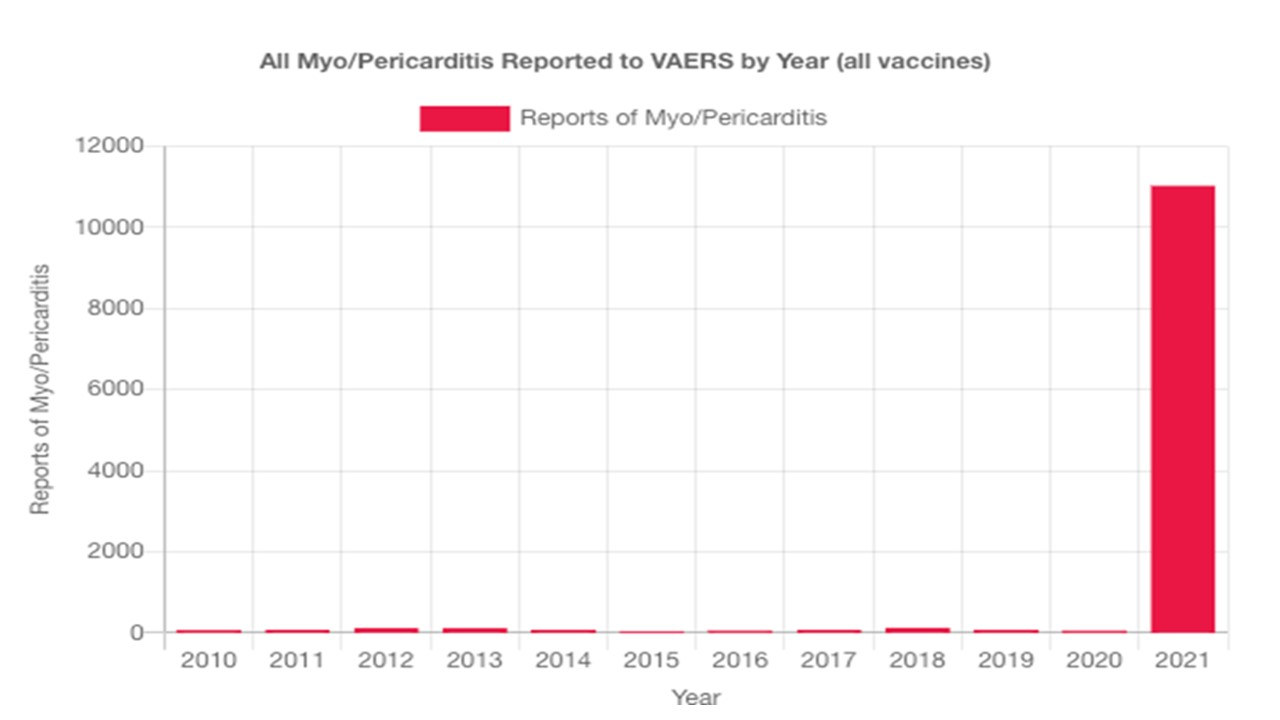
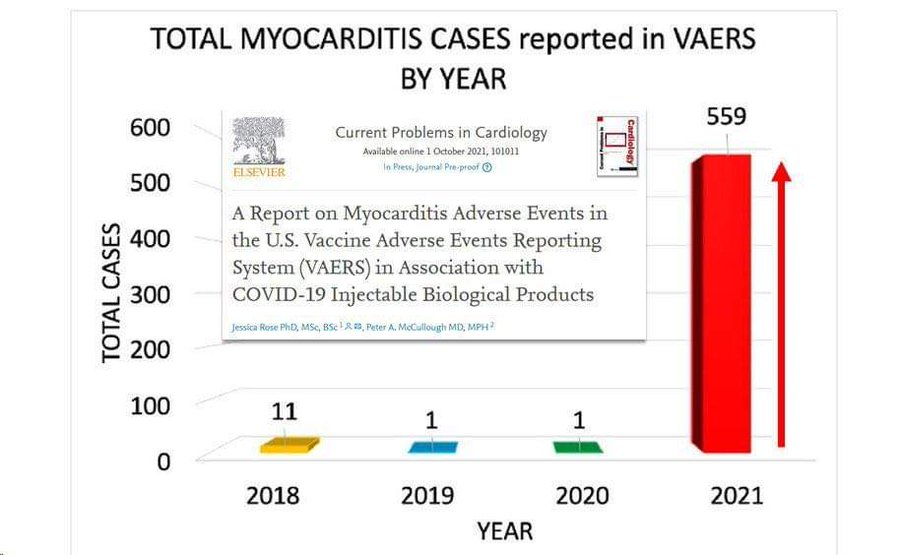
Myocardites associées aux injections antiCovid aux USA selon les données du VAERS
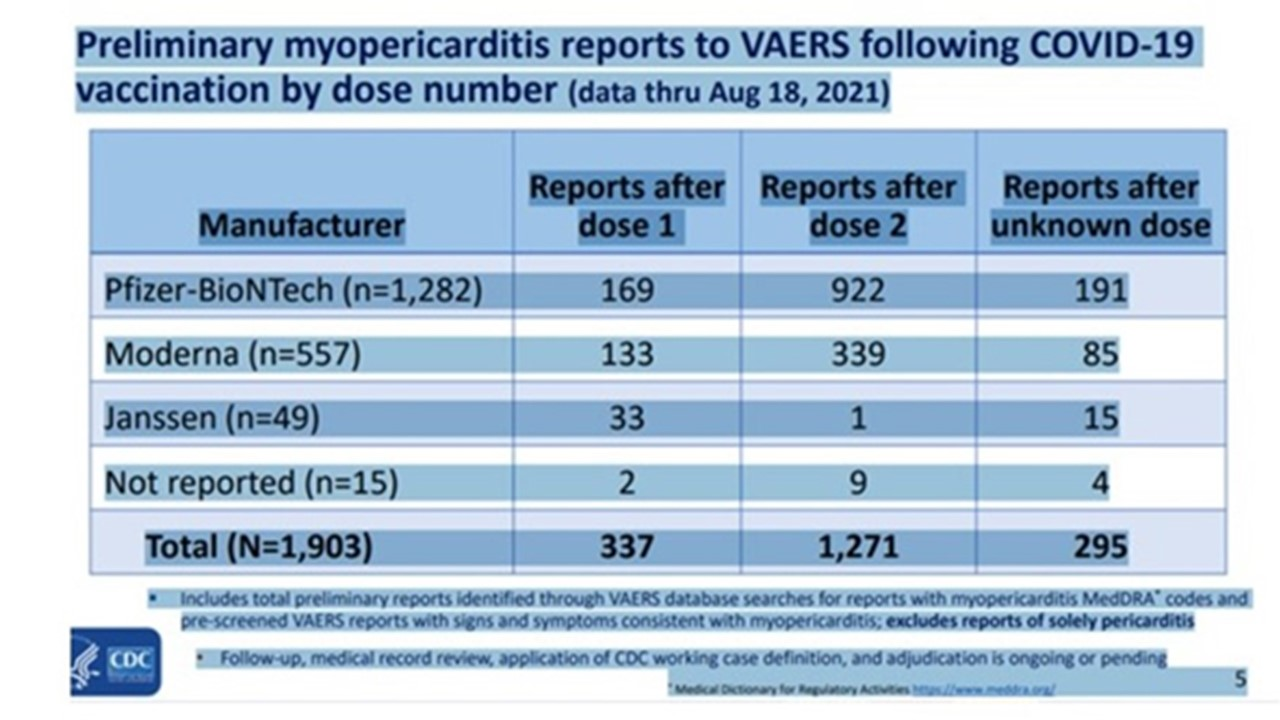
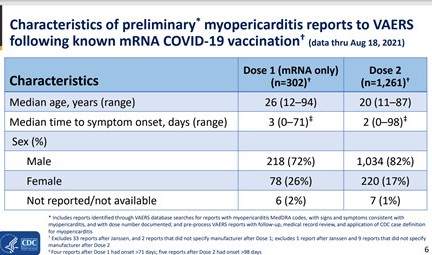
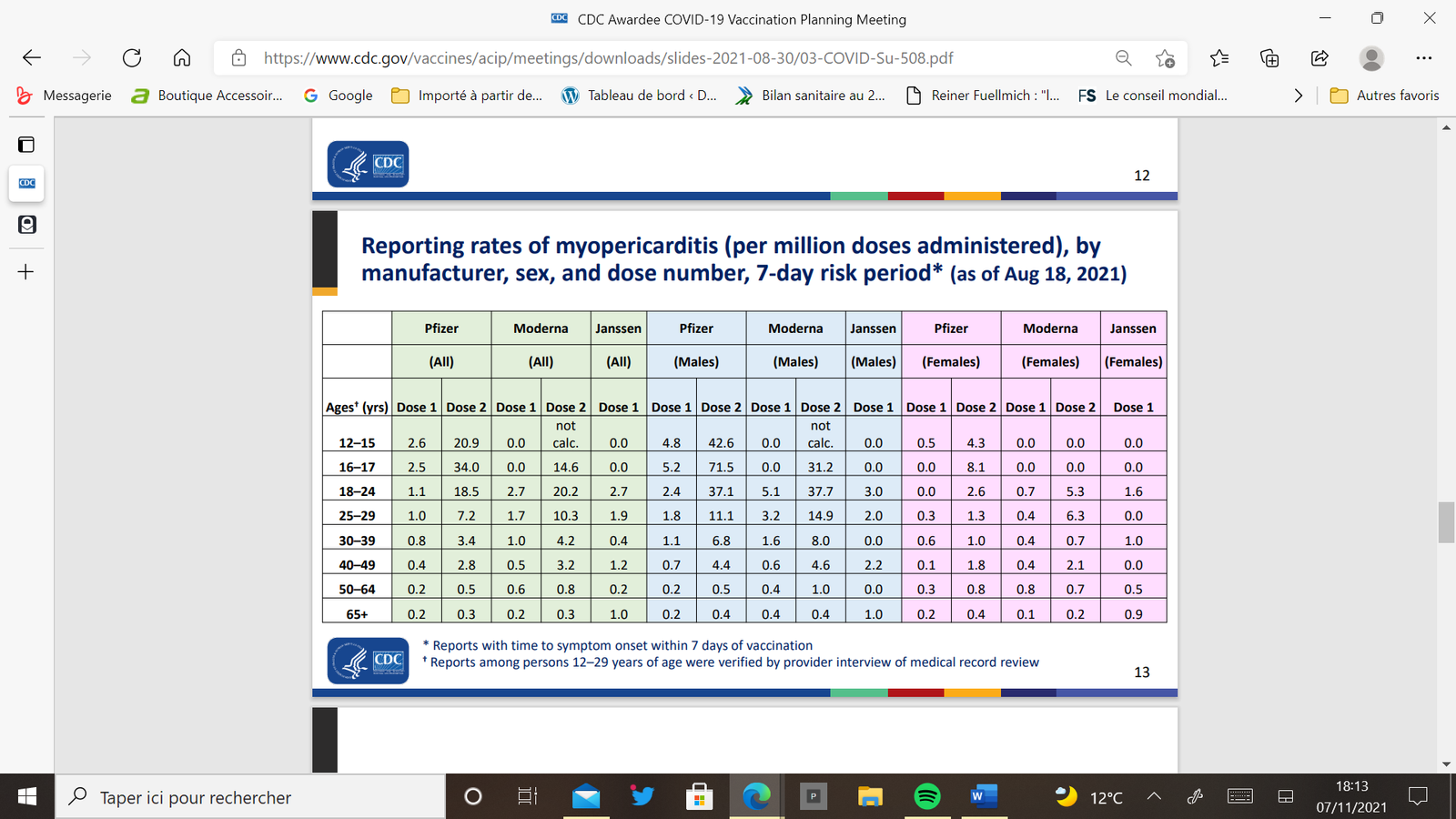
Les évènements indésirables cardiaques sont nettement plus fréquents chez les jeunes garçons d’autant plus qu’ils sont plus jeunes et qu’ils ont reçu deux doses de vaccin. Les signes cliniques apparaissent dans les jours suivant l’injection.
Relevons la fréquence considérablement augmentée chez les jeunes garçons par rapport à la population générale : 23 % de plus entre 12 et 15 ans, 40 fois plus entre 16 et 17 ans.[9]
Une autre étude dite stratifiée a été réalisée sur les myocardites chez les adolescents et leurs liens avec les vaccins à ARNm à partir des données du VAERS pour comparer le danger respectif de la maladie Covid et du vaccin.[10]
En se limitant aux seuls effets cardiaques (myocardites et péricardites en particulier), on peut conclure que la vaccination contre la Covid-19 chez les adolescents américains en bonne santé est plus dangereuse que le risque de la maladie elle-même
Le CDC rapporte qu’il y a 94 à 96% d’hospitalisation des myocardites/péricardites post-vaccinales.
Les garçons de 12 à 15 ans sont 12 fois plus sujets à des effets indésirables cardiaques que les filles du même âge, et cela principalement après la seconde injection du vaccin.
Pour les garçons de 12 à 15 ans, ont été enregistrés 162,2 cas d’effets indésirables cardiaques par million d’injections de vaccins anti-Covid, contre 94 cas / million chez les 15-17 ans, évènements ayant nécessité une hospitalisation.
Cette étude stratifiée du VAERS montre que le risque de myocardite avec troponine élevée chez les adolescents de 12 à 17 ans après vaccin est de 2,6 à 6,1 fois plus élevé que le risque d’hospitalisation à 120 jours pour la maladie Covid en août aux USA.[11][12]
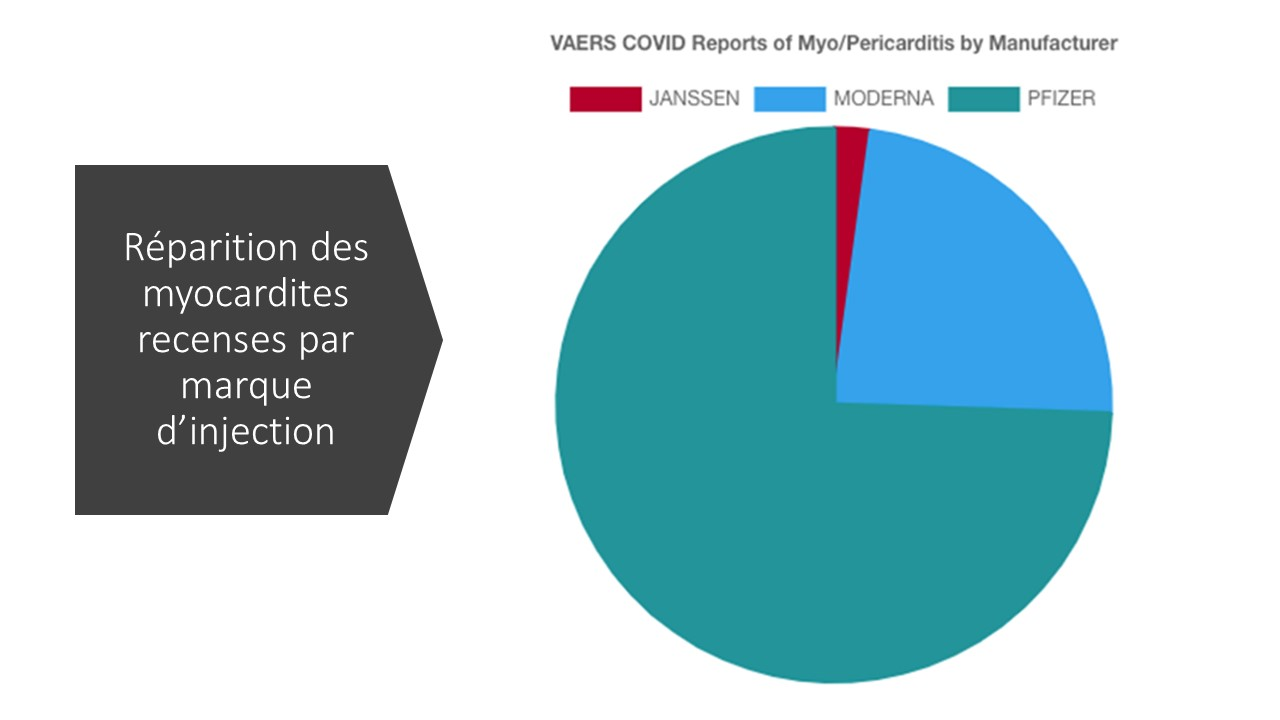
Comparaison de la fréquence générale des myocardites avant et après vax antiCovid
Une étude récente[13] chez l’adulte a comparé le taux de myocardites dans les périodes prévaccination et post vax[14] Jan 2019–Jan 2021) et pendant la période de vaccination Covid-19 vaccination (Fevrier-Mai 2021) au sein d’un large système de santé américain
Le nombre mensuel moyen de cas de myocardites ou de myo-péricardites pendant la période pré vaccinale était de 16.9 pour 100000 (95% CI, 15.3-18.6) vs 27.3 (95% CI, 22.4-32.9) durant la période vaccinale (P < .001).
Le nombre moyen de péricardites était pendant les mêmes périodes de 49.1 (95% CI, 46.4-51.9) et 78.8 (95% CI, 70.3-87.9), respectivement (P < .001).
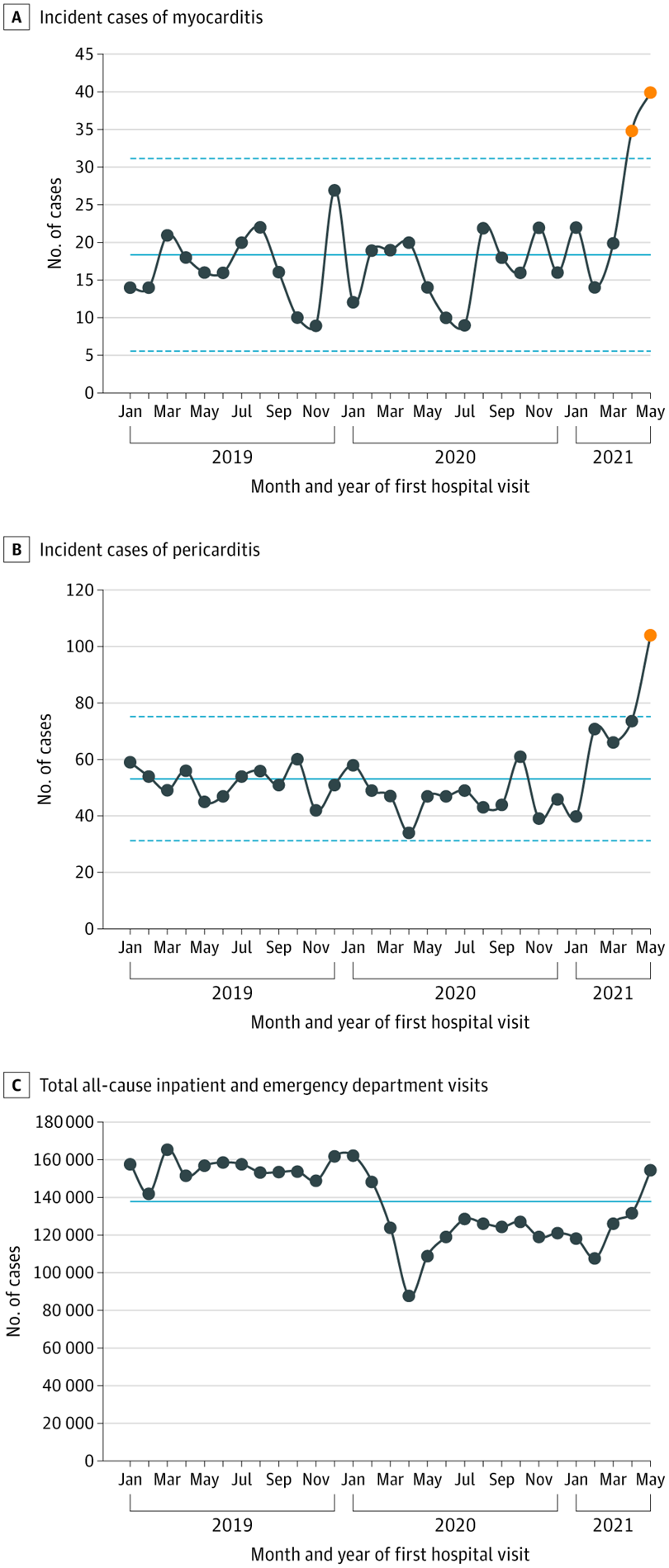
Les myocardites se développent rapidement chez les patients les plus jeunes et surtout après la seconde dose. Les péricardites affectent surtout les patients plus âgés et soit après la première, soit après la seconde.
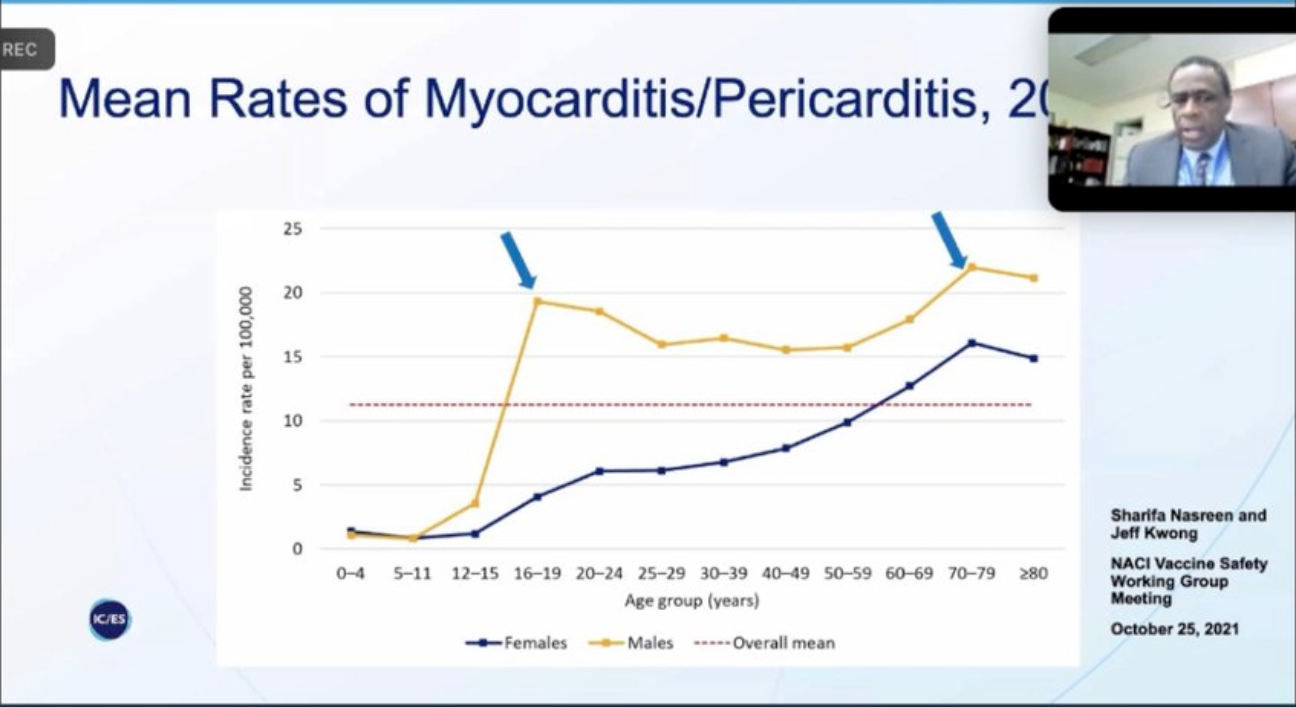
Les CDC américains (centres officiels : Centers for Disease Control and Prevention) ont rapporté l’association entre Covid-19 mRNA vaccins and myocardite surtout chez les garçons les plus jeunes dans les quelques jours suivant la deuxième injection.
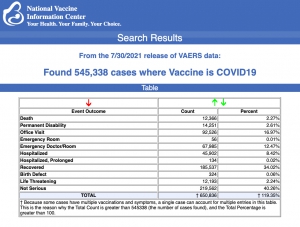
Sur la base des données du Vaccine Adverse Events Reporting System, le CDC a estimé que l’incidence de la myocardite après toute vaccination Covid-19 est de 0,48 cas pour 100 000 au total et de 1,2 cas pour 100 000 parmi les vaccinés âgés de 18 à 29 ans.
Un rapport de la campagne de vaccination de l’armée américaine contre le Covid-19 a noté une incidence de 8,2 cas de myocardite pour 100 000 hommes militaires (un total de 23 cas). Les enquêteurs de l’étude militaire ont identifié un dysfonctionnement ventriculaire gauche chez 17% des hommes (tous les cas étant légers ou légers à modérés).
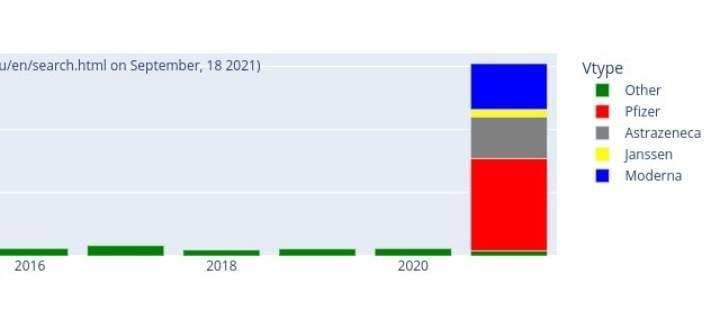
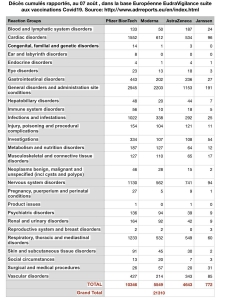
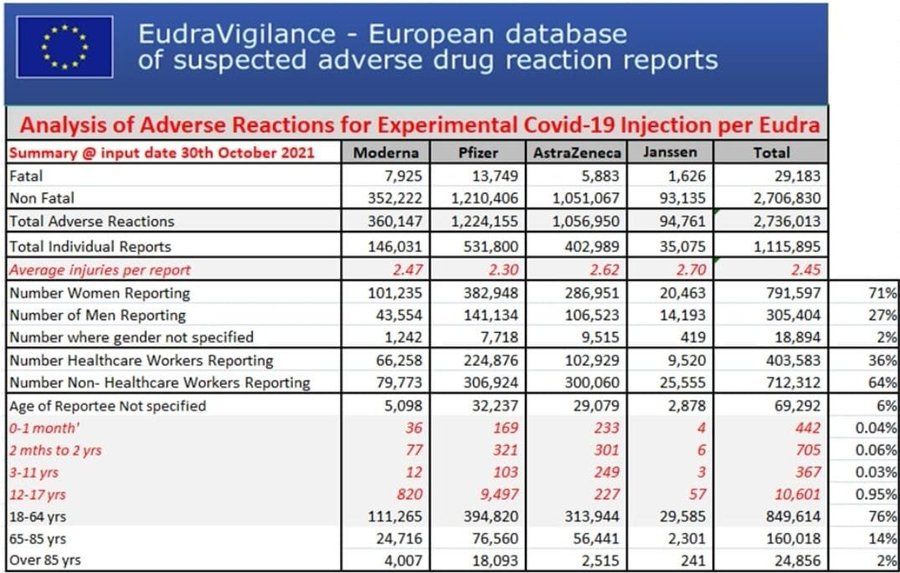
Myocardites en UE selon le site officiel de l’EMA : Eudravigilance
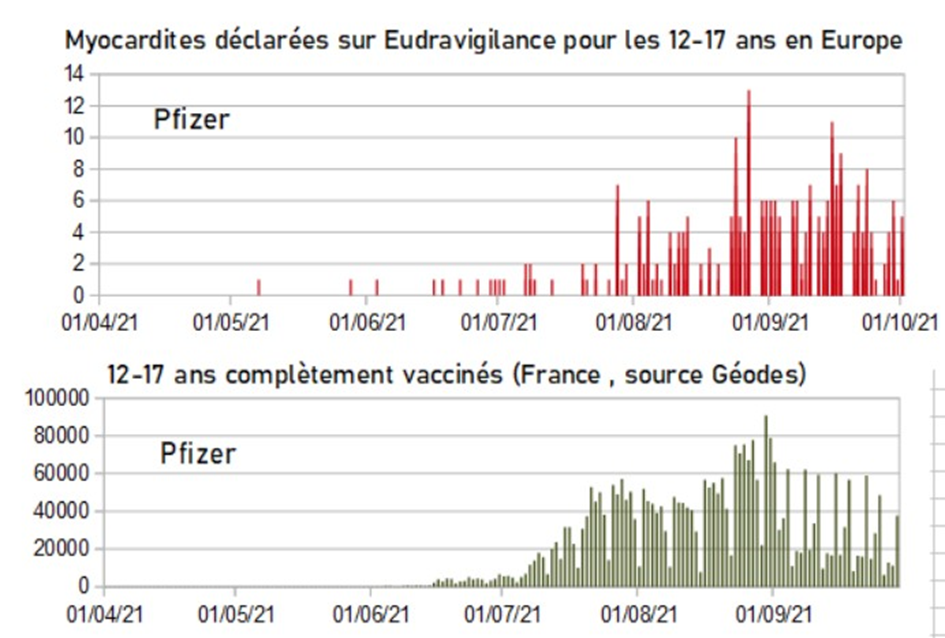
Myocardites des 12-17 sur Eudravigilance et vaccination des 12-17 ans en France
Du 1er avril au 15 juin, 3 myocardites ont été déclarées sur Eudravigilance chez les 12-17 ans. Puis le « vaccin » est arrivé pour les enfants, et le nombre de myocardites a explosé ! Alors, pure coïncidence ?[15]
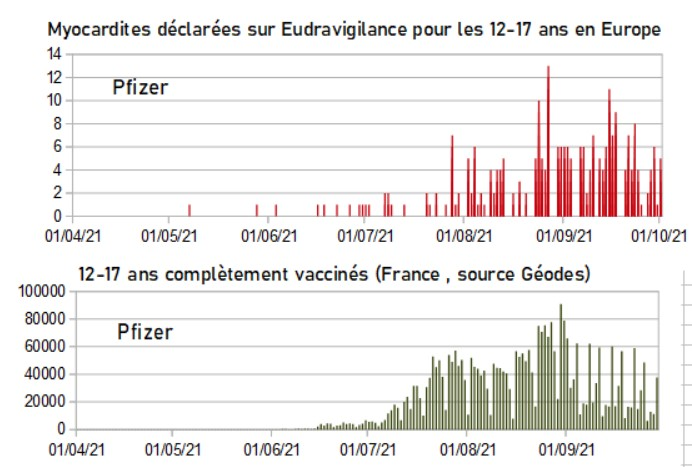
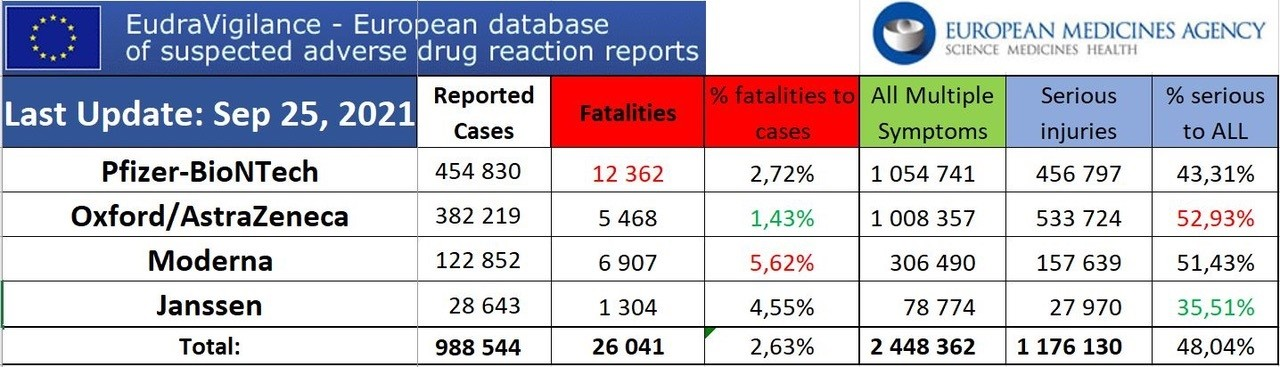
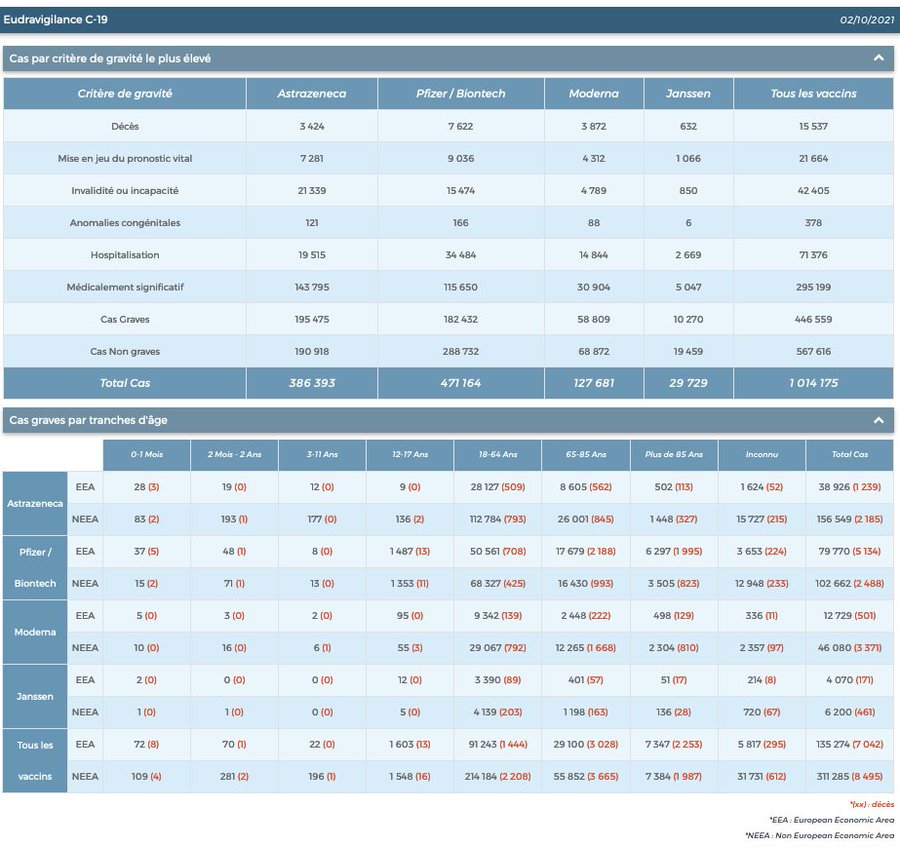
Eudravigilance (2.10.2021).
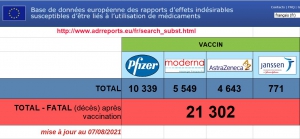
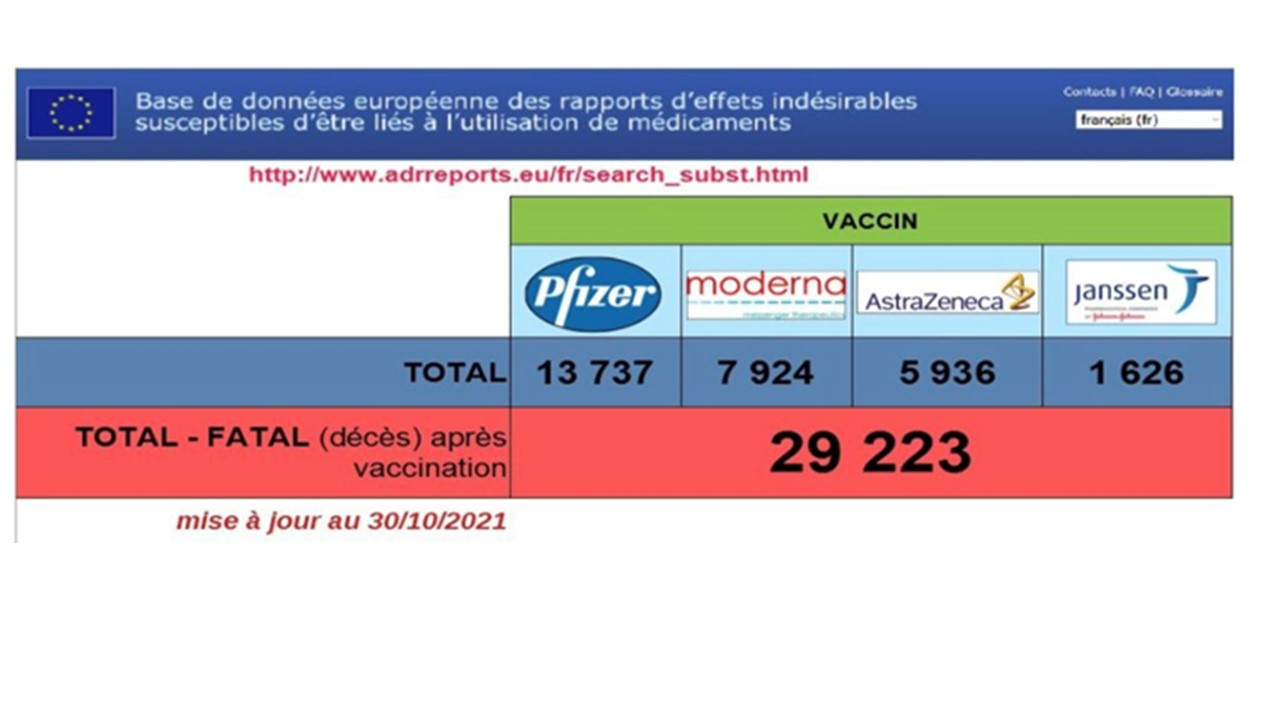
À ce jour, + de 28 000 décès liés aux « vaccins » déclarés dans les pays membres de l’UE.
Sur 12-17 ans, plus de 500 myocardites imputables aux Vaccins antiCovid.
Les Effets secondaires globaux dits pudiquement « indésirables » s’approchent des 3 millions.. Et l’on sait tous que les remontées d’informations sont inférieures à 5 %. Qu’en est -il de la réalité ?
Resituons les atteintes cardiaques dans les atteintes globales en UE (sur 27 pays hors G-B)
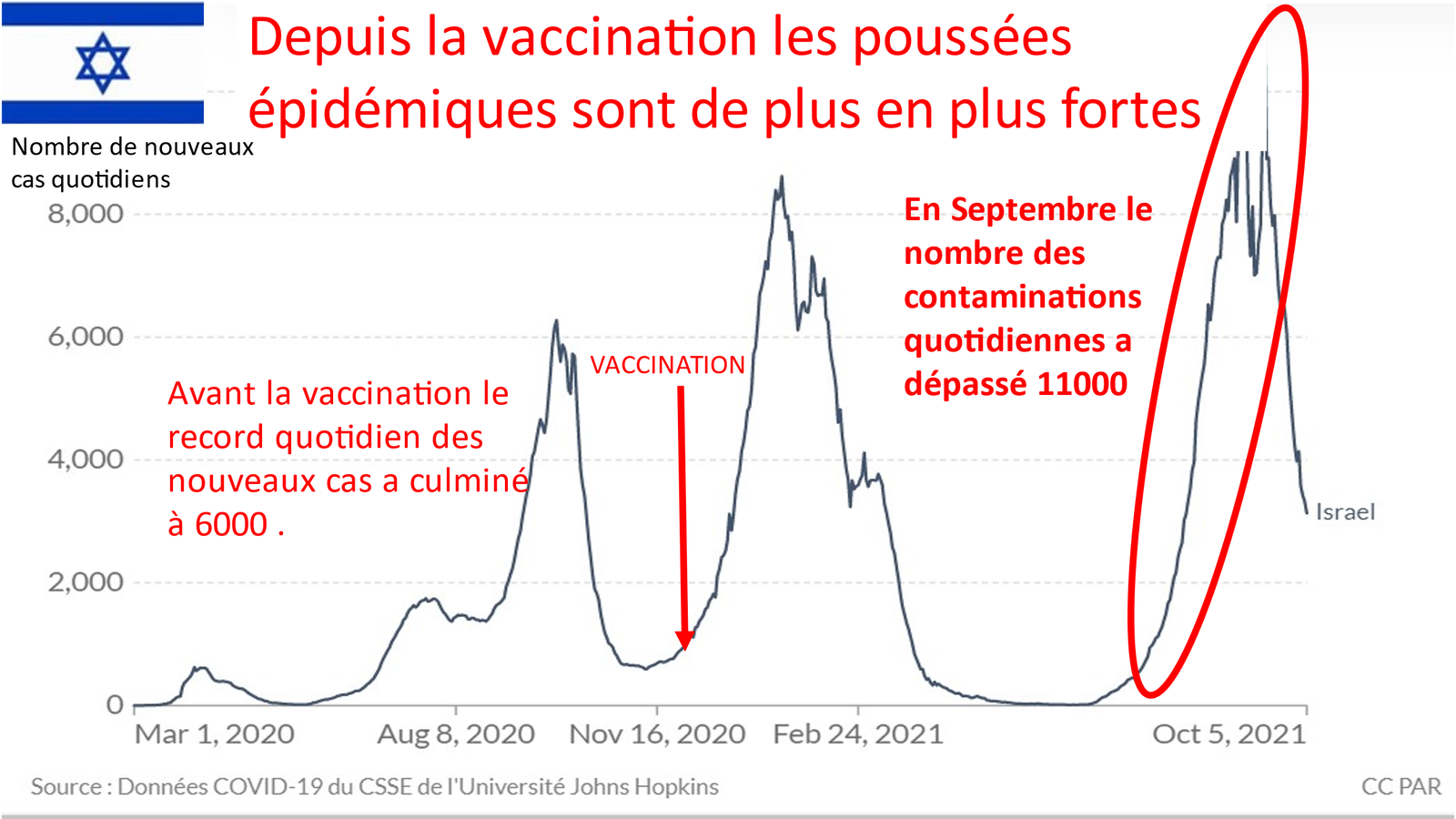
Les études israéliennes
Un premier rapport d’Israël en avril 2021 a mis en évidence une relation séquentielle apparente entre la réception du vaccin à ARNm Covid-19 et la myocardite, la plupart survenant chez des jeunes hommes auparavant en bonne santé et avec une incidence 5 à 25 fois supérieure au taux de fond habituel.
Le mécanisme précis reste obscur et les théories incluent un effet trophique direct du virus (bien qu’il n’y ait aucun rapport d’isolats viraux provenant d’une biopsie endomyocardique), et une réponse immunitaire exagérée de l’hôte invoquant les cellules T cytotoxiques en tandem avec la tempête de cytokines
En conséquence, la Food and Drug Administration aux États-Unis et l’Agence de la santé publique du Canada ont décidé d’ajouter des étiquettes de mise en garde aux vaccins à ARNm Pfizer et Moderna soulignant le risque de maladie cardiaque inflammatoire. Cela a été signalé secondairement par l’EMA et il a été demandé aux médecins de signaler ce risque … tout en ne dissuadant pas de faire l’injection…
Le rapport initial d’Israël note une incidence
de 1 sur 3000 à 1 sur 6000 myocardites
suite à la vaccination chez les jeunes adultes.
Les médecins israéliens ont été les premiers à sonner l’alarme dans un silence assourdissant dans les premières semaines.[16]
Ils ont évalué l’incidence de la myocardite après vaccin à ARNm BNT162b2 (Pfizer) dans une seule organisation de soins de santé (HCO) en Israël et décrit l’évolution clinique et la gravité de la maladie à partir d’un examen des dossiers des patients.
Les estimations du VAERS américain sont inférieures à celles israéliennes, probablement en raison des différentes méthodes utilisées pour identifier les cas (déclaration passive au CDC vs dossiers de santé électroniques en Israël ).
Dans l’étude israélienne de Barda et al.la vaccination a entraîné un excès de 2,7 cas de myocardite pour 100 000 personnes vaccinées.
Dans cette étude de cohorte rétrospective portant sur des personnes âgées de 16 ans ou plus dans un grand système de santé israélien, l’incidence estimée de la myocardite dans les 42 jours suivant l’injection (d’au moins une dose du vaccin à ARNm BNT162b2) était de 2,13 cas pour 100 000 vaccinés et 10,69 cas pour 100 000 chez les hommes âgés de 16 à 29 ans.
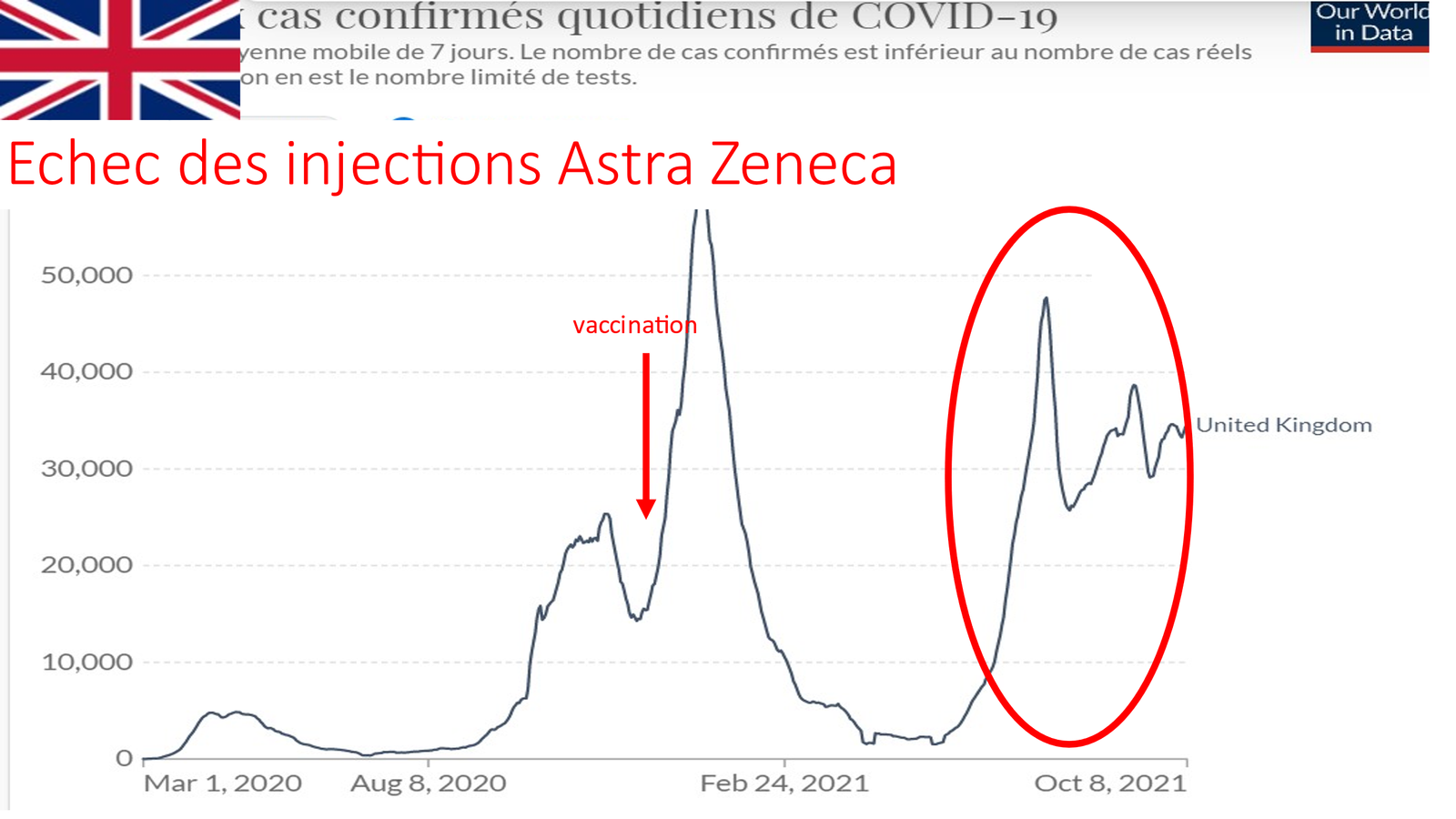
Les données récentes de l’office national de statistiques anglais sont (ONS) également très inquiétantes pour les enfants[17][18][19]
« Les données de l’ONS pour 2021 montrent qu’entre le 25 juin 2021 et le 17 septembre 2021, il y a eu 217 décès parmi les adolescents âgés de 15 à 19 ans, ce qui signifie que les décès chez les adolescents de plus de 15 ans étaient 47 % plus élevés qu’à la même période en 2020.
La différence ? L’augmentation du nombre de décès d’adolescents en 2021 a coïncidé avec l’offre du vaccin Covid-19, comme le montrent les données du NHS qui peuvent être téléchargées, et accessibles sur le site Web du NHS, montre qu’au 27 juin 2021 plus de 147 000 personnes de moins de l’âge sur 18 avaient reçu au moins une dose d’un vaccin contre le Covid-19 ».
L’Agence de réglementation des produits de santé et de médecine anglaise a ouvertement admis qu’elle soupçonnait la myocardite et la péricardite d’être des effets secondaires potentiels des vaccins Pfizer et Moderna Covid-19, en particulier chez les jeunes hommes.
UK Medicine Regulator a ajouté officiellement des avertissements concernant la myocardite et la péricardite aux étiquettes de sécurité des vaccins Covid-19. L’EMA également et les généralistes ont reçu des lettres les incitant à expliquer ce risque aux futurs vaccinés. Mais s’ils le font, qui se vaccinera ? Et si les médecins ne vaccinent plus, ils se font tancer par les ARS et l’ordre des médecins… (comme en France ou en Belgique et en suisse.)
Les données sont maintenant publiées pour que les autorités voient, une augmentation de 63 % des décès d’adolescents masculins depuis qu’ils ont commencé à recevoir le vaccin antiCovid-19, elles doivent enquêter sur cela et cesser immédiatement le déploiement de cette injection expérimentale aux enfants.
En Angleterre, la bataille continue ! Le mardi 5 septembre, Chris Whitty, le médecin-chef anglais a décidé d’annuler le Comité mixte sur la vaccination et de conseiller au gouvernement de proposer le « vaccin » anti-Covid 19 à tous les enfants du secondaire en bonne santé. Cela a jusqu’à présent conduit à augmenter de 400 % le nombre de décès chez les enfants de sexe masculin par rapport à la même période en 2020.[15]
Il y a environ 15,6 millions de personnes âgées de 19 ans et moins au Royaume-Uni, ce qui signifie que seulement 1 enfant et adolescent avec comorbidité grave, sur 410 526 serait décédé du Covid-19 en 18 mois. Seulement 1 enfant sur 1,7 million serait mort du Covid-19 en 18 mois, sans comorbidité connue.
Il n’y avait donc aucune excuse pour imposer cette injection dans les écoles et les journaux se demandent si le médecin-chef a compris qu’il mettait ainsi en danger la vie des enfants anglais.
Cependant, les gens se sont battus devant les tribunaux pour annuler la décision du médecin en chef de l’Angleterre[16] selon laquelle les enfants devraient recevoir une injection expérimentale de Covid-19, malheureusement en vain jusqu’à présent.
Un juge a ordonné au gouvernement britannique de soumettre des preuves justifiant la vaccination des enfants contre le Covid-19, leur donnant une date limite du lundi 11 octobre.
L’ordre de l’honorable juge Jay est le bienvenu après que la révélation, le jeudi 30 septembre, que depuis que les adolescents de plus de 15 ans ont reçu le vaccin Covid-19, les décès dans ce groupe d’âge ont augmenté de 47 % par rapport à la même période en 2020.
Mais l’examen par la Cour traîne depuis le premier septembre sans suspension des injections, et les drames se poursuivent…
En conclusion
1°) Très peu d’enfants sont malades (présentent des signes de maladies) du Covid (moins de 5% des malades recensés), ils ne font que des formes légères (trois fois moins de risque de mourir que de la grippe saisonnière). Ils ne peuvent pas espérer de bénéfice personnel de la vaccination.
2°) Les enfants ne constituent pas un vecteur de transmission. Les pseudo vaccins actuels ne protègent pas de la maladie et n’empêchent pas de la transmettre. On ne peut donc pas espérer que les vacciner puisse protéger les autres.
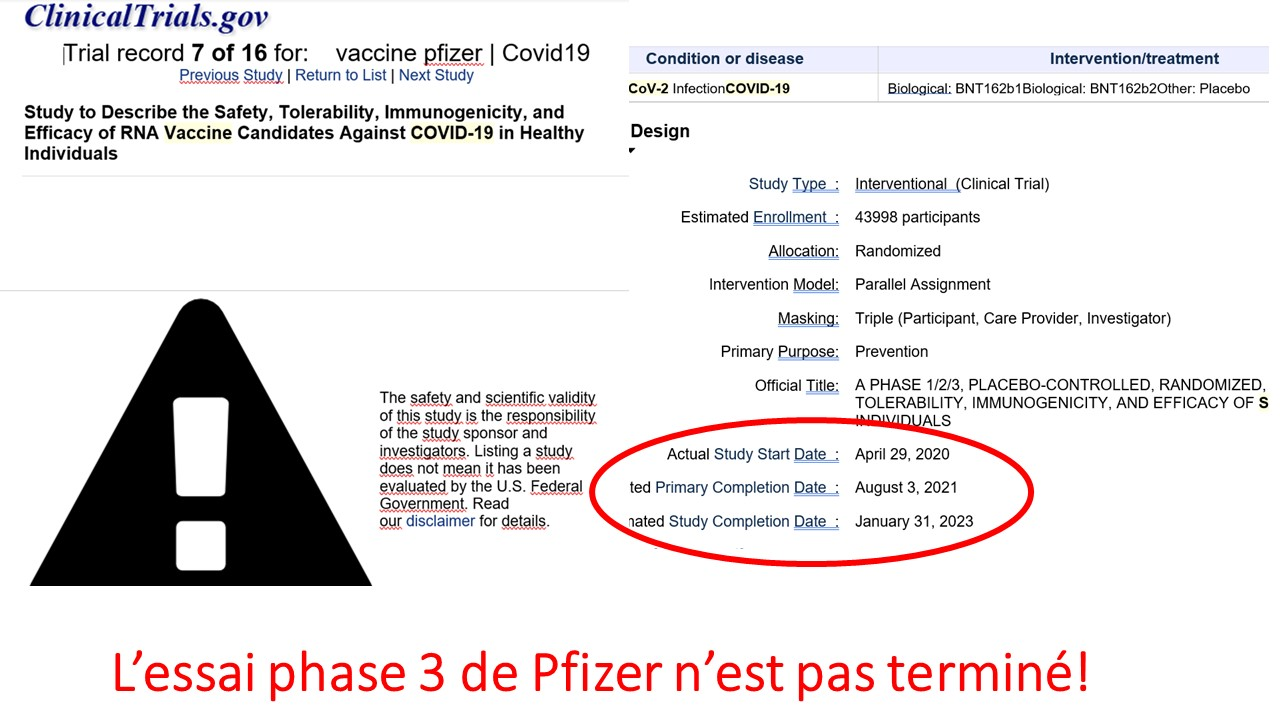
3°) les pseudo vaccins actuels, issus d’une technique jusqu’ici jamais utilisée en pathologie infectieuse humaine, sont totalement expérimentaux : les résultats des essais ne seront connus qu’en 2023. On ne connaît donc pas la totalité de leurs risques.
À court terme ils exposent à des complications sévères, dont des myocardites.
Aucun bénéfice, complications nombreuses et parfois mortelles la balance avantage risque est totalement défavorable chez l’enfant
ET si vous voulez revenir au concret, chez de vrais enfants et non des statistiques
Je terminerai par le récit fictif d’Alain Tortosa à propos d’un enfant sain de six ans brutalement décédé futur astronaute qui rejoindra plus vite que prévu les étoiles. Si des larmes ne vous montent pas aux yeux, demandez-vous ce que les mesures antiCovid ont fait de vous…[20]
J’avais une petite sœur Annie, un papa et une maman, des papis et des mamies et tout plein de cousins.
À l’école j’adorais jouer au super héros masqué avec les copains…
J’étais fier d’être enfin dans la grande école et je leur disais que j’avais 6 ans et demi quand on me posait la question.
Mais c’était un petit mensonge, car j’avais que 6 ans et 4 mois.
Papa et maman répétaient souvent que nous avions la chance de vivre dans une démo-cra-tie, ils disaient des mots que je comprenais pas très bien comme « nous sommes nés libres et égaux en droit »
Toute la famille était heureuse… mais ça, c’était avant et j’ai peur qu’ils soient plus jamais heureux.
Quand je serais grand, je voulais être astronaute et je savais pas que j’irais si vite dans le ciel au milieu des étoiles… à 6 ans.
Papa et maman, ils regardaient beaucoup la télé au repas du soir.
Cette méchante télé qui racontait tous les jours qu’on va tous mourir du méchant virus.
Ils étaient très content il y a quelques mois de se faire la piqûre pour battre le méchant virus Covid qui peut tuer tout le monde, même les petits enfants comme moi comme ils disent à la télé.
Moi je comprenais pas comment on peut être content quand le docteur il vous fait une piqûre.
Il y a quelques semaines à la télé, ils ont dit que les petits enfants aussi ils pouvaient maintenant être vaccinés.
Que le vaccin il marche très très bien et qu’il fait même pas mal.
Le président de l’Amérique, il a dit pour la vaccination des enfants de 5 ans :
« Un tournant dans notre bataille contre le Covid-19. »
et
« Cela permettra aux parents de mettre fin à des mois d’inquiétudes pour leurs enfants et réduira la proportion avec laquelle les enfants transmettent le virus aux autres.[1]»
Papa et maman m’ont dit que ce serait bien si moi aussi j’avais le vaccin comme les grands.
Mais moi je voulais pas parce que j’avais peur des piqûres.
Ils ont voulu me rassurer, « c’est rien, c’est comme une piqûre de moustique ».
Mais comme j’avais encore peur, ils m’ont dit que si je le faisais, je serais un super héros qui protège papi et mamie du super vilain méchant virus.
Je trouvais ça un peu bizarre parce que papi et mamie ils ont déjà le vaccin, la super méga armure pour se protéger du méchant virus comme ils m’avaient dit.
Et que en plus ils allaient encore recommencer pour être encore plus super protégés !
Papa et maman m’avaient répondu que j’étais trop petit pour comprendre et que j’aurais un diplôme de tueur de méchant virus.
Le jour du rendez-vous, j’avais quand même un peu peur, même si je faisais semblant pour pas rendre triste papa et maman.
Quand je suis arrivé au centre, tout le monde était gentil avec moi.
Ils trouvaient que j’étais super fort alors je gonflais mon corps pour leur montrer.
Avant la piqûre, on a vu un docteur qui a posé des questions à papa et maman.
Est-ce que j’avais des maladies, est-ce que mon petit cœur marchait bien, si il y avait des maladies dans la famille.
Papa et maman ont répondu et ont dit que j’étais un petit garçon super en forme sans maladies, que j’étais super sportif aussi.
J’aimais pas quand ils disaient que j’étais un petit garçon parce que maintenant j’étais grand.
J’ai dit au docteur que j’avais un peu peur.
Il m’a rassuré et il m’a dit que c’était rien que ça pouvait faire un peu mal après à l’endroit de la piqûre, mais c’est tout, et que ça pouvait pas faire peur à un grand garçon costaud comme moi.
Alors j’ai dit « d’accord je vais faire la piqûre pour tuer le virus et protéger papi et mamie »
J’ai vu que papa et maman ils étaient super fiers de moi et ça m’a encouragé.
Ça m’a fait presque même pas mal et après l’infirmière, elle m’a mis un pansement Spiderman.
Pour fêter ça, on est allés manger le soir au Macdo avec ma sœur et papi et mamie…
J’étais le héros de la soirée.
J’ai passé une super nuit à rêver d’étoiles.
Le matin, après un bon petit dej, papa m’a amené au foot comme tous les mercredis.
J’ai montré aux copains mon pansement Spiderman et ils ont trouvé que j’étais un super héros.
Mon copain Marcel m’a dit que j’avais de la chance d’avoir été vacciné, mais que ses parents ils voulaient pas, ils disaient que c’était dangereux.
Moi j’ai dit à Marcel que c’était des « complotistes » et des « chochottes ».
Le match a commencé, j’étais un peu fatigué, un peu comme si je venais de me réveiller,..
Mais je me suis donné à fond, le match était presque terminé…
Jean m’a fait une passe, j’allais marquer un super but et, au moment de tirer, j’ai senti mon corps m’abandonner, devenir tout mou et je suis tombé.
Je me sentais bien, j’avais pas mal et je comprenais pas pourquoi tout le monde courait vers moi.
D’abord les copains, puis l’entraîneur, puis papa !
Au bout d’un certain temps, je sais pas si c’est longtemps ou pas, une ambulance est arrivée avec des docteurs.
Ils avaient l’air super inquiets et je comprenais pas pourquoi…
J’ai eu comme un choc dans ma poitrine puis mes yeux se sont fermés, comme pour se coucher.
J’entendais les adultes crier, les copains pleurer, puis il y a eu un grand silence dans lequel on n’entendait plus que le bruit des oiseaux.
C’était comme si ils voulaient pas faire de bruit pour pas me réveiller.
C’est à ce moment que je suis sorti de mon petit corps.
J’avais l’air si bien, si détendu, si paisible, si innocent…
Papa était sur moi, il pleurait et je comprenais pas pourquoi il pleurait autant.
Je lui disais que tout va bien, mais il m’entendait pas…
C’est alors que maman est arrivée en hurlant, elle a tellement secoué mon corps que j’ai eu peur qu’elle me casse.
Les docteurs ont repoussé doucement maman, ils ont mis mon corps sur un lit à roulettes avec un drap blanc sur la tête et dans l’ambulance des pompiers.
À l’hôpital papa et maman, plutôt papa, parce que maman elle continuait à pleurer sans s’arrêter.
Papa a demandé ce qu’il s’était passé…
Les docteurs ont dit que mon petit cœur avait lâché d’un coup, que j’avais fait un inf, un infac, non un « infractus » si j’ai bien compris…
Papa avait l’air en colère et il a demandé pourquoi et le docteur il a dit que c’est très très rare, mais ça arrivait des fois, « un risque sur un million » qu’il a dit….
Papa a répété que j’allais bien, que j’avais jamais jamais eu des problèmes de cœur, que j’étais en bonne santé, que j’aurais pu vivre jusqu’à 100 ans et même plus !
Et le docteur il a redit que ça arrivait des fois, encore la semaine d’avant dans un autre club de foot, un jeune de 20 ans était mort comme moi sur le terrain sans qu’on sache pourquoi.
Papa il alors dit qu’il voulait savoir et il a demandé une « opopsie »… je crois que ça sert à regarder à l’intérieur du corps, ce qui a fait pleurer encore plus maman.
J’aurais tellement voulu faire un câlin à maman, mais j’avais plus de corps pour la serrer dans mes bras. J’étais très triste que elle pleurait sans s’arrêter comme si elle allait mourir aussi, mais de chagrin.
Plus tard, quand le résultat de « l’opopsie » est arrivé, le docteur il a dit à papa que j’avais fait une « miographie[2]» je crois, et que mon petit cœur avait pas supporté les efforts.
Je me rappelle maintenant que j’avais entendu une très vieille madame « ile » à la télé, une grande savante qui disait avec le sourire que c’est sûr que des enfants vaccinés feraient des « miographies », mais qu’en l’entendant c’était pas grave…
Que grâce à la vaccination de tous les jeunes enfants, le méchant virus il circulerait moins vite.
Et les journalistes et les docteurs de la télé ils étaient tous d’accord avec elle et contents !
D’ailleurs pour les docteurs de la télé, je suis comme ils disent une « statistique ».
Je suis un nombre si petit qu’on peut dire que c’est rien.
C’est bizarre, mais quand je regardais les yeux de papa et maman à l’église, devant mon petit cercueil blanc, j’avais pas l’impression que c’est rien pour eux et que j’étais une « statistique ».
Voilà, c’est l’heure, je dois partir, je voulais être un astronaute, je voulais aller dans les étoiles et c’est fait…
Je vais rejoindre les autres enfants, les autres « statistiques » qui sont maintenant au ciel.
Grâce à eux, leurs papis et mamies sont protégés du méchant virus et mon papi et ma mamie aussi.
J’espère qu’ils sont fiers de moi, que maintenant ils peuvent vivre à l’abri du virus.
Alain Tortosa[3]
5 novembre 2021 https://7milliards.fr/tortosa20211105-je-m-appelais-albert-j-avais-6-ans.pdf
- [1] https://www.france24.com/fr/am%C3%A9riques/20211103-Covid-19-aux-%C3%A9tats-unis-la-campagne-de-vaccination-des-enfants-de-5-%C3%A0-11-ans-lanc%C3%A9e
- [2] https://sfcardio.fr/actualite/risque-de-pericardite-ou-de-myocardite-apres-vaccin-arnm-contre-la-covid-19
- [3] « À tous les enculés qui participent sciemment et froidement au crime d’enfants. »
Alors avant de croire des recommandations mercantiles prônant l’injection anti Covid à vos enfants souvenez-vous des faits indiscutablement établis.
- [1] https://theCovidworld.com/florian-dagoury-world-record-holder-in-static-breath-hold-freediving-diagnosed-with-myopericarditis-after-pfizer-vaccine-possible-end-of-career/[↩]
- [2] A partir de 13 ans! Longue liste d’athlètes « soudainement » décédés ou gravement malades (report24.news) [↩]
- [3] Veuillez signaler d’autres cas ou erreurs: redaktion@report24.news[↩]
- [4] R Vasudeva, P Bhatt, C Lilje Tendances des hospitalisations pédiatriques liées à la myocardite aiguë aux États-Unis, 2007-2016 AM J Cardiology . 15 juin 2021; 149:95-102 doi: 10.1016/j.amjcard.2021.03.019. Publication en ligne du 20 mars 2021.[↩]
- [5] Définition del’OMS maladie orpheline si incidence = ou < à 6 /100 000 habitants[↩]
- [6] Risk of Myocarditis from Covid-19 Infection in People Under Age 20: A Population-Based Analysis (nih.gov) [↩]
- [7] https://resistance-mondiale.com/vaccination-contre-larnm-Covid-19-et-developpement-de-la-myopericardite-confirmee-par-cmr[↩]
- [8] Le CDC, Centre for Disease Control, organisme officiel américain qui surveille les maladies aux USA. Le VAERS (Vaccine Adverse Event Reporting System) système officiel de pharmacovigilance américain.[↩]
- [9] CDC Awardee Covid-19 Vaccination Planning Meeting[↩]
- [10] https://www.medrxiv.org/content/10.1101/2021.08.30.21262866v1 « SARS-CoV-2 mRNA Vaccination-Associated Myocarditis in Children Ages 12-17: A Stratified National Database Analysis »[↩]
- [11] SARS-CoV-2 mRNA Vaccination-Associated Myocarditis in Children
Ages 12-17: A Stratified National Database Analysis »
https://medrxiv.org/content/10.1101/2021.08.30.21262866v1.full.pdf[↩] - [12] Vaccination plus risquée que la Covid-19 pour les 12-17 ans – REINFOCovid[↩]
- [13] Forty hospitals in Washington, Oregon, Montana, and Los Angeles County, California, that were part of the Providence health care system and used the same electronic medical record (EMR) were included[↩]
- [14] George A. Diaz, MD1; Guilford T. Parsons, MD, MS2; Sara K. Gering, BS, BSN3; et al Audrey R. Meier, MPH4; Ian V. Hutchinson, PhD, DSc5; Ari Robicsek, MD2 Myocarditis and Pericarditis After Vaccination for Covid-19 JAMA2021 Sep 28;326(12):1210-1212.doi: 10.1001/jama.2021.13443
Myocarditis and Pericarditis After Vaccination for Covid-19 – PubMed (nih.gov) [↩] - [15] Yoann 3 oct sur tweeter[↩]
- [16] Myocarditis after Covid-19 Vaccination in a Large Health Care Organization
Guy Witberg, M.D., Noam Barda, M.D., Ph.D., Sara Hoss, et all
Myocarditis after Covid-19 Vaccination in a Large Health Care Organization | NEJM. Oct 21[↩] - [17] Augmentation de 63 % des décès chez les adolescents au Royaume-Uni — Morts après vaccination (wordpress.com) [↩]
- [18] Investigation : Deaths among male Children have increased by 400% since Chris Whitty decided they should have the Covid-19 Vaccine – The Expose[↩]
- [19] UK Judge orders Government to provide evidence in court that justifies Covid-19 Vaccination of Children in legal challenge to halt roll-out of the jabs to Kids – The Expose[↩]
- [20] Je m’appelais Albert, j’avais 6 ans et demi… – Nouveau Monde (nouveau-monde.ca) [↩]