IL FAUT DECONFINER RAPIDEMEMENT : économie et santé des 67 millions de français en jeu
Confinement aveugle de la population, mesure illogique, inefficace, dangereuse et ruineuse
Passons à la méthode coréenne et/ou allemande
(Cet article date du11 avril 2020 et les chiffres fournis sont ceux du 10/4/2020 10 heures GMT.)
Par Gérard Delepine chirurgien oncologue, statisticien et Nicole Delépine pédiatre cancérologue
La seule stratégie qui a prouvé qu’elle était efficace pour arrêter une épidémie est de fermer précocement les frontières, de dépister massivement, puis de confiner les contaminés et/ou les traiter, tout comme les cas à risques. La population non contaminée doit pouvoir se protéger (masques, gel) et poursuivre ses activités pour permettre à la nation de disposer de toutes les armes nécessaires pour combattre.
Le confinement aveugle que Macron nous impose n’est-il qu’un écran de fumée destiné à masquer l’état misérable de nos hôpitaux, dont il est en partie responsable et ses erreurs initiales de gestion de crise ? Le confinement aveugle est-il destiné à accoutumer la population à la dictature ? Cette mesure moyenâgeuse, médicalement inefficace est toxique pour les confinés et économiquement catastrophique[1] [2] .
Le président Macron a déclaré la guerre ! Mais à qui ? Au virus ou aux français ? Et avec quelle stratégie ?
L’histoire semble se répéter.
En 1940, l’ennemi était allemand ; aujourd’hui c’est un virus. Mais dans les deux cas, des gouvernants, incompétents se sont montrés imprévoyants, inefficaces, menteurs, et rejetant ensuite sur nos compatriotes la responsabilité de l’échec de leurs décisions désastreuses
En décembre et janvier 2020, alors que le Covid19 tuait allègrement les chinois, le gouvernement a parié que ce virus épargnerait la France comme le célèbre nuage de Tchernobyl et a passé son temps à minimiser le danger.
Pourtant le caractère nouveau du virus retrouvé chez un patient chinois dès octobre 2019 était reconnu, et l’imprévisibilité de son évolution évidente. Il n’a pas préparé le pays contre la menace qui s’annonçait. Il n’a pas renforcé les hôpitaux et en particulier les services d’urgence et de réanimation pourtant en grève depuis 11 mois pour crier leur misère. Il n’a pas reconstitué le stock de masques dispersé par une ministre précédente, ni fait fabriquer des tests, ni accumulé les quantités de désinfectants et gels hydro alcooliques nécessaires à la lutte. Il n’a pas embauché de personnel soignant. Il n’a pas commandé de respirateurs alors que leur nombre était notoirement très insuffisant. Et lorsque les chinois ont indiqué que la chloroquine était efficace contre le virus, la ministre n’en a pas fait fabriquer ; elle a interdit par décret sa vente sans ordonnance alors que celle-ci durait depuis 50 ans sans signalement particulier d’accident toxique.
L’orthodoxie financière européenne et le dogme mondialiste dictaient leurs vérités : la France ne disposait pas d’argent magique et de toute manière, il ne pouvait venir que de bonnes choses de l’étranger.
En mai 1940, notre état-major a été incapable de défendre nos frontières, mais au moins il a essayé. En février 2020 la France a perdu la bataille des frontières parce le gouvernement ne l’a pas livrée. Et lorsque l’ennemi s’est installé en France, Pétain comme Macron ont utilisé cette défaite pour justifier la suppression les droits élémentaires du citoyen. Coup d’état sanitaire sans aucune justification médicale sérieuse rendu possible par des députés godillots ou tétanisés par une peur irrationnelle créée de toute pièce par une propagande massive des médias complices.
S’agit-il d’incompétence ou d’une mise en scène Machiavélique (tu régneras par la peur) destinée à imposer la dictature au service de la finance ? Les déclarations de l’ex-ministre Buzyn mettent en doute leur seule incompétence. Ils savaient depuis fin décembre et ils n’ont rien fait pour contrer l’épidémie. Et les premières lois votées en « urgence sanitaire » ont supprimé les dernières protections sociales (durée hebdomadaire du travail) héritées de la résistance et de de Gaulle.
Aujourd’hui Macron bloque l’essentiel de l’activité économique du pays sans protéger efficacement ceux qu’il envoie au front. Pas de masque pour les médecins de ville dont déjà au moins 6 ont payé de leur vie cette stupidité. Pas de masque pour les policiers et gendarmes qui vont contrôler des ausweis que même les nazis ne demandaient pas de jour[3]. Pas de masques pour les ouvriers lorsqu’ils prennent les transports en commun bondés, haut lieu de contamination. Et la porte-parole du gouvernement a longtemps publiquement nié leur utilité, alors que leur emploi était unanimement recommandé par tous les experts étrangers. Le gouvernement a refusé d’en commander jusqu’au 27 mars, avec plus de 80 jours de retard et de mensonge sur leur utilité !
Mais peut être fallait-il de nombreux morts et une campagne de propagande prolongée pour que la panique s’installe dans la population et qu’elle accepte l’état d’urgence et la dictature sans se soulever ?
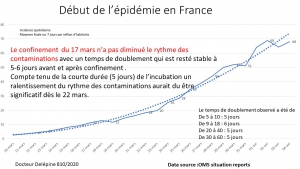
Le confinement aveugle (sans dépistage) de la population n’était pas nécessaire[4]
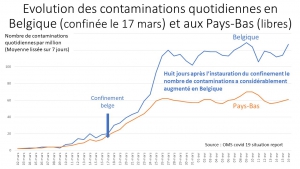
Pour stopper ou ralentir une épidémie, le contrôle aux frontières, la quarantaine des malades et des contacts à risque reconnu grâce au dépistage généralisé, l’hygiène publique [5]et privée[6], l’utilisation de moyens de protection constituent les méthodes qui ont fait leur preuve en stoppant la plupart des pestes historiques[7] sans l’aide d’antibiotiques ni de vaccin. Le Japon, Taiwan, la Corée du Sud, et Singapour ont réussi à limiter très fortement la dissémination du Covid19 sans assigner à résidence de la population. Leur stratégie a été suivie avec succès par l’Allemagne et d’autres pays du nord de l’Europe.
Au Japon, la bataille des frontières a été spectaculairement illustrée par la quarantaine du paquebot Diamond Princess et de ses 3711 passagers et membres d’équipage, parmi lesquels on comptera plus de 625 contaminés et 7 morts. Elle a été complétée par l’isolement des personnes à risque, des mesures ciblées de restriction de transports et d’entrée sur le territoire et la généralisation des gestes barrière par la population dont l’utilisation de masques, la fermeture des écoles et le report des manifestations publiques, en particulier sportives.
Aucune fermeture d’administrations, ni confinement généralisé de la population n’a été envisagé. Le bilan actuel de cette politique est actuellement infiniment plus satisfaisant que ceux des pays qui ont confiné aveuglément leurs populations. Avec une population double de celle de la France ou de l’Italie, et le triple de celle de l’Espagne,
Le Japon ne comptait au 10/4 que 5347 infectés et 88 morts
Contre respectivement 85351 infectés et 12192 décès en France, 143626 contaminés
Et 18281 morts en Italie, et 152446 infectés et 15238 morts en Espagne, 24923 contaminés et 2523 morts en Belgique tous pays européens apôtres du confinement aveugle de leur population[8].
En Corée du Sud, la guerre des frontières contre le Covid19 a été menée vigoureusement. Elle a permis, sans confinement de la population, de limiter la contamination à 10450 cas et à la mortalité à 208 coréens (pour 51 millions d’habitants) dont une grande majorité a été infectée par les membres d’une secte religieuse qui revenaient de Wuhan et l’avaient caché. Et la vie là-bas se poursuit quasi normalement alors que nous français sommes tous punis, astreints à résidence inutilement [9].
A Singapour, les mêmes mesures ont permis sans confinement de la population de ne compter que 1910 contaminés et 7 morts parmi ses plus de 5,8 millions d’habitants.
A Taiwan, dès le 31 décembre 2019, les Centers for Disease Control de Taïwan ont mis en œuvre des mesures d’inspection pour les vols en provenance de Wuhan. Le 20 janvier 2020, le gouvernement a activé le Centre de commandement central des épidémies, qui a mobilisé des fonds gouvernementaux et du personnel militaire pour faciliter la production de masques faciaux en envoyant dès février des soldats dans les usines des principaux fabricants de masques pour doter en personnel les 62 nouvelles lignes de production de masques. Les autorités taïwanaises ont suspendu les visites en Chine à partir de février 2020. Taiwan a enregistré la prévalence de covid19 par habitant la plus basse du le monde (0,2/100000).
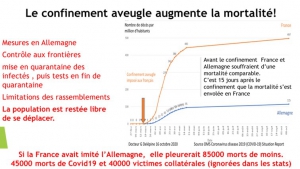
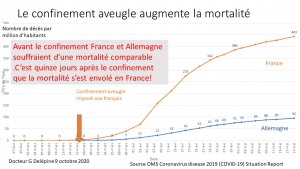
L’Allemagne a suivi la stratégie coréenne : tests massifs, quarantaine pour les personnes à risque, géolocalisation et délivrance de certificats d’immunité pour permettre à ceux déjà immunisés de reprendre une activité normale. Ses résultats au 10 avril (2373 morts en Allemagne contre 18281 en Italie) confirment que cette stratégie sans confinement aveugle est la plus efficace aussi en Europe.
Ces exemples démontrent que le confinement aveugle des populations décrété par les dirigeants totalitaires n’est pas nécessaire lorsque les mesures traditionnelles contre les épidémies sont utilisées.
Pire : Le confinement aveugle de toute une population a été inefficace
Dans l’histoire des épidémies, le confinement de la population saine n’a jamais apporté la preuve de son efficacité en situation réelle. Ce sont l’hygiène et la quarantaine sélective des malades, et seulement des malades et de leurs contacts à risque qui ont aidé à éradiquer la peste, la lèpre, le typhus ou le choléra.
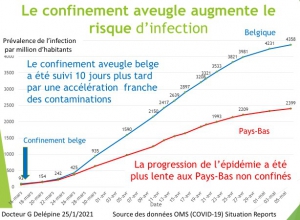
Les pays (Italie, Espagne et France) qui ont les premiers imposé le confinement aveugle à leur population sans tester, ni traiter ont enregistré les plus grands nombres de contaminés. Pour s’en convaincre il suffit d’étudier la prévalence[10] du covid19 selon les chiffres de l’OMS au dernier relevé connu (le 10 avril 2020).
L’Italie avec une prévalence de 237/100 000 et l’Espagne prévalence de 326/100 000 sont aux premières places des contaminés suivies par la Belgique (215/100 000), l’Allemagne (prévalence 135/100 000) et la France (prévalence 130/100 000). Les pays asiatiques qui ont appliqué strictement les méthodes classiques de dépistage, quarantaine des malades et moyens de protection sans confinement global aveugle ont tous une prévalence inférieure à 50 (Japon 42, Singapour 32 et Corée 20).
L’Italie que notre gouvernement a choisi malheureusement comme modèle, l’Espagne et la Belgique, qui ont aussi imposé un confinement aveugle constituent des exemples de résultats calamiteux. Elles comptabilisent les plus fortes prévalences mondiales de la maladie et les mortalités record (30/100 000 pour l’Italie, 32/100 000 pour l’Espagne et 22/100 000 pour la Belgique), alors que les pays européens qui ont privilégié des quarantaines ciblées ont des mortalités en moyenne dix fois inférieures (2/100 000 pour la Norvège, 3/100 000, pour l’Autriche, 2,8/100 000 pour l’Allemagne.
Et comme le confinement italien s’est révèle catastrophique, son gouvernement le prolonge [11] sans plan de sortie défini pour l’instant. Le coté inhumain de son application (armée dans les rues, fortes amendes lors des enterrements, peines de prison) laissera probablement de lourdes séquelles psychiques chez de nombreux confinés. Choisir un tel modèle pour la France fait preuve de beaucoup d’improvisation, sauf si le but réel était de saisir l’opportunité d’un conditionnement de la population pour imposer la suppression des libertés publiques.
Le confinement d’une population entraîne de lourdes complications médicales
Les complications médicales et sociales de cette mesure moyenâgeuse inefficace, sont avérées et souvent catastrophiques. Tous les confinés interrogés décrivent un sentiment d’isolement et le manque de contacts sociaux. L’absence de tout contact physique avec les membres de la famille et amis confinés ailleurs, est particulièrement mal vécu surtout lorsque le confinement aveugle est prolongé.
On achève bien les chevaux … et maintenant l’hécatombe programmée de nos ainés.
Pour les vieillards, pensionnaires des EHPAD, la solitude complète imposée par le confinement les conduit très vite au « syndrome de glissement » et à la mort, presque aussi sûrement que le virus. Quelle ineptie et quelle honte de leur interdire les visites de leurs proches, alors que leurs soignants sortent chaque jour et prennent des transports publics bondés et reviennent un jour ou l’autre porteurs du virus qu’ils propagent à leur insu car scandaleusement peu protégés et rarement testés. Et que dire de l’interruption des rééducations pour dysphagie nécessaires à beaucoup des pensionnaires ?
Et que penser d’une note du ministère aux directeurs d’EHPAD précisant clairement que les personnes âgées malades ou susceptibles de l’être ne doivent en aucun cas être adressées aux urgences car elles ne seront ni hospitalisées, ni traitées, ni réanimées. Précisons qu’il s’agit de critères d’âge absolu, sans tenir aucun compte de l’état physique et intellectuel des résidents. Nous connaissons tous de beaux vieillards de plus de 90 ans et des « vieux » de 70 ans ou moins. Les médecins ont toujours pesé ces éléments du temps où les docteurs pouvaient exercer leur vocation selon Hippocrate, et non sous l’autorité malfaisante des bureaucrates interdisant d’hospitaliser les infectés de plus de 70-75 ans et précisant qu’ils ne doivent pas être admis en réanimation pour ne pas encombrer les lits de réanimation « inutilement ».
Les nazis à la descente du train de déportés triaient selon l’âge physiologique. Pas nos bureaucrates. Les plus de 70 ans infectés sont renvoyés dans leurs EHPAD ou faute de possibilité d’isolement réel, de traitement, de personnel et de matériel de protection ils contaminent leurs compagnons d’infortune sans même pouvoir dire adieu à leurs familles. Nos technocrates ont-ils osé imaginer un début de solution au financement des retraites par le génocide des plus vieux retraités, selon les conseils de J Attali, mentor du président ?
Les récits dramatiques se succèdent sur les réseaux sociaux. Couples déchirés dans leurs derniers moments, enfants qui ne reverront jamais leur mère, soignants qui veillent depuis des mois ou années sur des vieillards et les voient brutalement contaminés, refusés d’hôpital, interdits de réanimation, privés de traitement, abandonnés à la mort sans personne pour leur tenir la main puis jetés au tombeau en catimini[12], sans accompagnement, les familles recevant des contraventions de 135 euros s’ils se retrouvent pour les accompagner au cimetière. Avec l’épidémie de décès dans les EHPADs, elles risquent même la prison pour récidive d’infraction en attendant peut-être qu’on les condamne à mort comme l’Antigone de Sophocle, emmurée vivante pour avoir bravé l’interdiction royale d’enterrer son frère. Eternel combat de la morale qui définit notre humanité contre des lois tyranniques injustifiés[13]
Pourquoi devrions-nous attendre la fin de l’épidémie pour réagir[14] ? C’est maintenant que nous devons nous soulever pour stopper cette monstruosité, ce génocide de nos ainés.
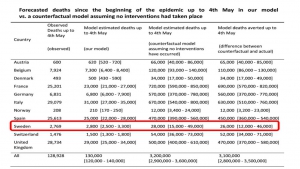
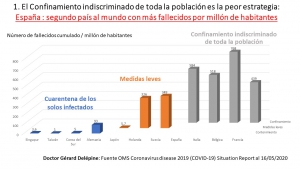
Et pendant cet enfermement imposé, que chacun d’entre nous joigne le maximum d’élus, maires, conseillers régionaux et députés, sénateurs pour qu’ils transmettent au gouvernement la réalité des chiffres montrant que le confinement aveugle général s’accompagne de beaucoup plus de morts que l’isolement ciblé des malades et de leurs contacts.
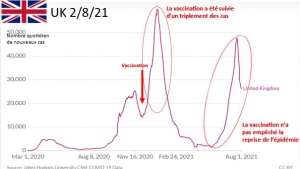
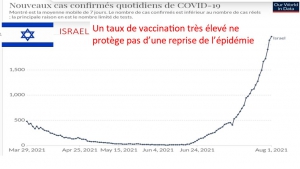
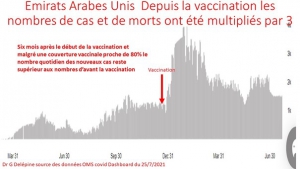
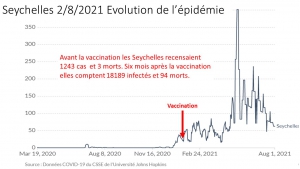
Envoyez-leur le diagramme joint comparant les taux de mortalité.
Mais les plus jeunes ne sont pas épargnés
Les complications sanitaires du confinement sont nombreuses et bien connues [15]: dépressions qui persistent dans plus d’un tiers des cas de nombreux mois après sa levée, stress post-traumatique durable, obésité, drames familiaux, divorces, retard scolaire, addiction renforcée à l’alcool aux drogues ou aux écrans.
Certains groupes de personnes sont encore plus à risques. Enfants placés, qui se retrouvent encore plus seuls avec la moitié des encadrants absents. Femmes et/ou enfants victimes de violence familiales confinées avec leurs bourreaux eux-mêmes agacés par l’absence de sortie et paniqués par le Covid19. SDF dont la première mesure anti Covid19 fut de leur mettre des contraventions parce qu’ils trainaient dans la rue … UBU roi au royaume de France. [16]
Prisonniers et malades des hôpitaux psychiatriques privés de visites, malades attendant interminablement aux urgences pour accidents vasculaires, domestiques, ou professionnels et qui pour la plupart n’ose plus aller « encombrer les urgences, laissant évoluer bien des symptômes.
Malades chroniques priés d’attendre avec renouvellement de leurs ordonnances par le pharmacien… La consultation médicale serait donc devenue inutile ? Les généralistes, trop souvent dupes de l’intérêt du confinement total devant les contaminations qu’ils constatent (en raison de la honteuse absence prolongée de masques et de chloroquine) commencent à redouter les dégâts sur leurs patients abandonnés. La télémédecine tant vantée par les énarques désincarnés ne peut remplacer le contact humain direct avec le malade.
Les jeunes accouchées sont, elles aussi privées de la présence du père et de la visite de leurs proches. Période pourtant anxiogène pour les jeunes femmes particulièrement en période de psychose publique organisée.
Les adolescents privés de leurs premières amours sont complètement déprimés, le téléphone et même Skype et/ou face time, Instagram ou WhatsApp ne pouvant en rien remplacer l’intimité qu’ils commençaient à découvrir. Même leur école et leurs professeurs en chair et en os qu’ils critiquaient souvent leur manquent cruellement.
Les conséquences mentales sur des personnes saines habituellement, parfois psychologiquement fragiles et persuadées d’être confrontées à un danger de mort imminente, sont importantes et seront souvent durables dans le temps…La durée de la quarantaine est significativement liée à l’augmentation des symptômes dans toutes les études. Le nombre des personnes décompensées va s’accroître avec la durée du confinement et le renforcement quotidien de l’hystérie et de la psychose entretenues par le ministère de la santé, le président ou son premier ministre et diffusées servilement par les médias appartenant à quelques milliardaires amis du président.
« L’histoire de l’invocation de mesures de quarantaine est ternie par des menaces, une peur généralisée, un manque de compréhension, des discrimination, difficultés économiques et rébellion (…).la mise en quarantaine peut créer de graves problèmes psychologiques, émotionnels et financiers pour certaines personnes ». Et conclusion de l’article cité : « Nos données montrent que la mise en quarantaine peut entraîner une détresse psychologique considérable sous la forme de syndrome de stress post-traumatique et de symptômes dépressifs. Les responsables de la santé publique, les médecins spécialistes des maladies infectieuses et les psychiatres et psychologues doivent être informés de ce problème »[17] Cette étude ne porte pourtant que sur la quarantaine des personnes malades qui, de ce fait comprennent le bien-fondé de leur isolement… Les séquelles de l’assignation à résidence aveugle des populations italiennes, espagnoles et françaises (moins conditionnées à la dictature que la Chine) n’ont pas fini d’être analysées. Bel avenir pour les psychologues et les psychiatres.
Le confinement aveugle, catastrophe économique et sociale.
Ce confinement aveugle de la population est catastrophique pour le pays conduit tout droit vers une récession grave. Sans preuve d’efficacité médicale réelle, Il ruine l’économie. Son cout direct prévisible est actuellement estimé à 3% de notre Produit Intérieur Brut mais sa prolongation qui tente nos dirigeants pourrait faire passer l’addition à 5 ou 6% du PIB[18], soit près de 150 milliards ! Son coût indirect est encore plus lourd. Il menace déjà à court terme l’existence même des artisans, professions libérales, petits commerçants soumis à de nombreuses charges fixes (loyers, salaires, charges sociales) qu’ils ne pourront plus honorer faute d’activité. Il menace aussi leurs nombreux salariés jetés au chômage[19]. Sans oublier les travailleuses du sexe réduites au chômage total sans indemnités et les mendiants privés de toutes ressources. Après la fin de l’épidémie les populations vont donc souffrir davantage et bien plus longtemps des conséquences économiques du confinement (perte de salaire, chômage, chute du PIB, aggravation de la misère) que de la maladie.
Les raisons du confinement : les prédictions d’une boule de cristal anglaise qui s’est déjà lourdement trompée lors de l’épidémie de grippe H1N1.
Le scénario catastrophe qui aurait convaincu E. Macron de mettre toute la Nation en prison n’est pas une analyse objective de résultats médicaux avérés, mais une simulation, une prédiction[20]. Dans la Grèce antique, les rois consultaient l’oracle de Delphes avant de prendre des décisions importantes. Le 12 mars 2020 Macron a consulté Neil Ferguson, un épidémiologiste de l’Imperial College à Londres qui aurait prédit de 300 000 à 500 000 morts en France en cas d’absence de mesures d’endiguement[21]. Comme dit le PR Raoult, ce n’est pas de la science, plutôt de la science-fiction qui évoque les prédictions de Nostradamus.
Nous avons déjà expliqué l’incertitude de pareilles prédictions en médecine[22] et leurs conséquences délétères, lorsque les hypothèses de départ sont fausses. Et les hypothèses retenues, très incertaines à cette date, du fait de la mauvaise connaissance de cette maladie nouvelle, se révèlent maintenant grossièrement fausses. Le traitement par ordinateur, s’il donne un aspect moderne à la prédiction, est incapable d’améliorer la pertinence des résultats. Si E. Macron s’était davantage intéressé à la réalité médicale et en particulier aux résultats des mesures prises au Japon, à Taiwan et en Corée du sud ou à Singapour, il aurait pu nous éviter ce cauchemar inefficace, inutile, toxique et hors de prix du confinement aveugle. Mais comme beaucoup de son entourage, il croit à la start-up nation, à la technologie, aux sciences exactes qui permettent d’aller dans la Lune, mais ignore que soigner un être humain est plus complexe et non réductible à un modèle. La médecine est un art mâtiné de science, mais pas une science exacte.
Erreur stratégique que n’a pas commise l’Allemagne. Erreur que nous payons maintenant[23] de plus de 12192 morts, alors que l’Allemagne avec une population 20% plus importante en déplore cinq fois moins (2373).
Bilan de 75 jours de combat de l’épidémie en France et de 26 jours de confinement
Frontières grandes ouvertes avec affichettes dans les halls d’aéroport comme arme de dissuasion massive contre un virus qui malheureusement ne savait pas lire le français…Le gouvernement Macron a laissé arriver de Chine et d’Italie tous ceux qui le voulaient, sans aucun dépistage à leur arrivée et encore moins de quarantaine imposée aux ressortissants des pays infectés et aux voyageurs suspects à l’arrivée. Il a fallu des critiques publiques répétées sur le manque de détection aux arrivées aéroportuaires pour que la direction générale de la santé annonce (très tardivement) la mise en place d’une « équipe médicale d’accueil » à l’aéroport de Roissy (sans tests).
Ce sont les syndicats du personnel aérien d‘Air France qui ont imposé d’arrêter la desserte des pays à risques. De plus, aucune protection des personnels par masques n’était organisée ! Le virus a pu s’installer tranquillement dans le pays.
Une « guerre » effectuée sans moyens logistiques, sans masques pour tous et partout, sans tests avec, pour seule arme, un confinement aveugle
La bataille de France est encore livrée par des soldats de première ligne en nombre insuffisant et peu armés. Toujours trop peu de masques et de tenue de protection pour les soignants et les personnels d’urgence et de sécurité, tests diagnostic toujours très rationnés, nombre de lits de réanimation indigne, (plus de trois fois inférieur à celui du Japon, de Taiwan et deux fois inférieur à celui de l’Allemagne)[24], nombre de respirateurs très insuffisants qui nous oblige à quémander des places chez nos amis allemands et suisses qui ont heureusement mieux géré la crise (et sans confinement généralisé). Et impossibilité pour les médecins traitant de prescrire de la chloroquine lorsqu’elle leur paraît indiquée.
Au 24 mars 2020, la France n’avait réalisé que 101 046 tests contre 500 000 tests par semaine en Allemagne, la Corée du Sud, ayant elle réalisé près de 400 000 tests depuis le début de l’épidémie. En rapportant le nombre de tests à la taille de la population nationale, le nombre de tests réalisés pour 10 000 habitants dans chaque pays, la France en réalise pour l’instant 15, l’Allemagne 80 et la Corée du Sud 77.
Aucune réouverture de lits d’hôpitaux, Aucune embauche de personnels soignants[25] ni revalorisation de leurs salaires pour les encourager à ne pas démissionner. Pas d’alignement du prix de consultations des médecins de ville sur la moyenne européenne rémunérations (> 40 €) malgré l’ampleur de la catastrophe des hôpitaux déjà en grande crise avant l’arrivée du virus. Les « héros » doivent continuer à travailler dans des conditions dignes de la brousse et se contenter d’applaudissements chaque soir à 20 H. Et notre système sanitaire surnommé Titanic dans des tribunes et des manifestations n’en finit pas de couler dans l’indifférence des dirigeants qui prodiguent flatteries et promesses, mais dont on attend toujours des actions concrètes.
Les héros sont fatigués d’être méprisés, mis en danger, jamais écoutés
Le président de la fédération des médecins de France, généraliste, JP Hamon lui-même touché par la maladie, insiste sur les médias : « nous ne sommes pas de la chair à canon ». Sans effet, en dehors de câlinothérapie du genre « vous êtes des héros ». Non, nous médecins ne sommes pas des héros, mais souhaitons faire notre métier dans des conditions de sécurité correctes pour nous et nos malades, ce qui nous est refusé.
Médecins et malades sans protection autres que celles qu’ils se fabriquent eux même ! En 1915, pour ramasser les blessés sur les tranchées de Verdun les soldats étaient munis de casque. Mais en 2020 en France, le Gouvernement a décidé de nous priver du matériel de protection de base. Et déjà au moins six médecins sont morts. Pourquoi ? Ils se moquent de nous, ou jouent à la guéguerre avec notre peau et par conséquence avec celle des patients qui nous consultent, privés de masques également. Dans quel but ?
Des policiers traités comme de la chair à virus, longtemps interdits de masque ! Envoyés à la quête de la fameuse attestation, contaminante potentielle, sans masque, et pire longtemps sans autorisation d’en porter même fabriquée par leur épouse ! il était « interdit de se protéger ». Pourquoi ? Le virus serait-il dangereux pour le monde entier sauf pour les policiers français ?
Les travailleurs méprisés
Ils doivent continuer à travailler selon les injonctions du ministère de l’économie, prendre des transports en commun bondés (lorsque la fréquentation diminue la RATP diminue le nombre de ses rames et la promiscuité reste égale), servir les clients en supermarchés pendant plusieurs semaines sans masques qui étaient inutiles pour eux comme le rappelaient chaque soir les autorités !
Le plus sidérant est l’acceptation apparente de la population tellement manipulée qu’elle finit par croire aux mensonges du pouvoir et accepter une mesure humiliante, inutile, inefficace et toxique. Soumission….Est-ce un test de notre résistance à la soumission, qui rappelle cruellement les expériences de Milgram [14] montrant qu’une grande proportion d’une population normale devient capable, sous l’ordre d’un chef, de se comporter en bourreau et de délivrer une dose mortelle d’électricité. Le sujet de Milgram (Monsieur tout le monde) agit contre ses convictions et en souffre. Cependant, il va se conformer à ce qu’on attend de lui ».[26]
Le confinement aveugle est une très mauvaise idée sur le plan sanitaire. Mais le véritable projet ne serait-il pas de poursuivre la destruction des acquis de notre modèle social sans manifestation grâce au confinement et I ’installation d’un état policier sur le modèle chinois ?
En 1940, la défaite aux frontières a servi de prétexte à une prise de pouvoir par le régime de Vichy, et à la suppression des droits fondamentaux des français. En 2020, le président, utilisant l’invasion virale, nous impose un couvre-feu diurne et nocturne, et veut finaliser la suppression des acquis du pacte social hérité de la Résistance et du CNR[27] ?
La population souffre d’une double peine ; menacée du virus, elle est de plus soumise obligée de montrer un « ausweis », un laisser passer de jour lorsqu’elle veut sortir. Pire que sous Pétain. Sous l’occupation allemande, aucune attestation sur l’honneur disant qu’on va promener son chien ou chercher son pain n’était exigée[28]. Ce confinement a d’ailleurs entraîné un exode des parisiens plus important que celui de 1940 ! Et des concitoyens arrivent à le leur reprocher, voire à les dénoncer, répétant ainsi les comportements des périodes les plus sombres de notre histoire[29].
« En ce joli mois de mars 2020, on est heureux d’apprendre que l’Etat chinois, jusqu’ici présenté comme une infâme dictature antidémocratique, peut soudain être pris pour modèle, qui plus est dans une disposition qui foule aux pieds les libertés publiques fondamentales. Ainsi meurt la liberté dans son pays de naissance, dans l’indifférence générale. »[30]
Et la loi d’exception votée le jeudi 19 Mars 2020 pour soi-disant lutter contre l’infection comporte la suppression de droits sociaux[31] et des restrictions de liberté qui laissent craindre le pire.
Ne vous laissez pas manipuler par le harcèlement quotidien de la propagande du gouvernement relayée par ses fidèles pseudo-experts et ses journalistes en continu sur les médias.
La volonté de protéger la nation serait plausible, si les confinés ne se retrouvaient pas sans protection au marché ou chez le boucher et pire aux urgences, car la vie continue et les infarctus et blessures n’ont pas disparu Et si ce confinement était « de bonne foi » pourquoi interdirait-on aux cyclistes de rouler seuls, aux cavaliers de sortir seuls, aux baigneurs de profiter du soleil seuls sur un coin de plage à distance des autres confinés, à un surfeur de pratiquer, et aux marcheurs de maintenir leurs formes, aux sportifs de continuer à s’entraîner seul[32] pour ne pas perdre des années d’exercice, aux plaisanciers d’aller avec leurs familles confinées avec eux, sur les mers…
Il y a une volonté d’humiliation des confinés qui démontre, s’il en était besoin l’absence de bon sens sanitaire et de projet réel de santé. En quoi un promeneur seul en campagne à plus de 500 m de chez lui est-il dangereux et justifie-t-il un rappel à l’ordre par un drone ?
Vous pouvez multiplier les exemples. Fermer les plages, les parcs est une mesure coercitive non médicale visant à conditionner le peuple à obéir, à se soumettre. Test pour voir jusqu’où un dictateur peut aller trop loin ?
Nos dirigeants veulent-ils détruire le pays par la gravité de la récession engendrée par le confinement, et ce dans le cadre d’une volonté mondiale de mise au pas par un gouvernement Mondial revendiqué encore en 2017 par le célèbre mentor du président Macron, Jacques Attali qui attendait une grande pandémie pour y parvenir. Nous y voici.
Etablir une dictature à la chinoise grâce à la panique et l’hystérie entretenue par la propagande sans précédent[33] que diffusent en continu les médias pour une infection dont la mortalité mondiale réelle reste bien inférieure à celle la grippe[34] ? Chaque soir, messe quotidienne oblige, vous apprenez le nombre de morts par coronavirus, mais on oublie de vous dire que 500 personnes sont parties de leur cancer ce même jour (et miracle si on leur trouve un coronavirus, ce dernier sera déclaré responsable du décès et probablement plus côté en tarification à l’activité) et la psychose est alimentée par des chiffres absolus sans jamais de comparaison à l’ensemble de la population de plus de 65 millions de français.
Pour mettre en perspective les chiffres absolus répétés chaque soir : rappelons quelques chiffres
« En 2013, 567 078 décès domiciliés en France entière (hors Mayotte) ont été enregistrés. Avec respectivement
163 602 et 142 175 décès, les tumeurs et les maladies de l’appareil circulatoire constituent les causes de décès les plus fréquentes, tous sexes confondus. »[35]
L’OMS estime que la grippe saisonnière fait 60.000 morts par an en Europe. En France, la grippe saisonnière touche chaque année en moyenne 2,5 millions de personnes et en tue environ 10000. Lors de l’épidémie grippale 2014-2015, la surmortalité hivernale a atteint 18.300 décès. (AFP février 2020). Alors pourquoi cette hystérie pour une maladie certes nouvelle, mais qui n’a tué en 2 mois (au 10/4) 12192 personnes ?
Ecoutons le Pr marseillais[36] Raoult sur le confinement et aussi la lueur d’espoir thérapeutique par le Plaquenil qui lui a valu des menaces, comme toujours avec les novateurs qui pourraient menacer des lobbies.
« Confiner l’ensemble de la population sans dépister et sans traiter, c’est digne du traitement des épidémies des siècles passés, et à peu près aussi inopérant.
La seule stratégie qui fasse sens est de dépister massivement, puis confiner les positifs et/ou les traiter, tout comme les cas à risque puisque c’est possible, comme on le voit en Chine et en Corée. »
Méditons l’analyse pertinente de Valérie Bugault[37]:
« L’analyse de la crise actuelle révèle aussi l’application des techniques du pompier pyromane et du triangle de Karpman, c’est-à-dire le jeu de rôles bourreau/victime/sauveur. Le pouvoir laisse la situation de crise s’installer, voire l’aide à s’installer, volontairement ou involontairement. Dans le réel, le pouvoir occupe donc la place du pyromane, donc du bourreau. Puis, une fois que la crise est installée et en cours, le pouvoir se présente comme le sauveur, qui va donc nous sauver de la crise qu’il a lui-même installé furtivement, tel un pompier qui éteindrait l’incendie après l’avoir lui-même allumé discrètement. »
Comme nos aïeux contre l’occupant nazi et ses alliés de Vichy, RESISTONS à la tyrannie pseudo médicale ! Exigeons des masques et des dépistages par des tests diagnostic généralisés et la chloroquine qui protègeront bien mieux la population que le confinement. Refusons le confinement aveugle qui détruit la vie sociale et l’économie sans bénéfice sanitaire réel. Refusons des lois d’exception injustifiées. RESISTONS !
[1] D’après AFP : En Chine depuis le confinement, la production industrielle a connu un repli de 13,5% sur un an, contre +6,9% en décembre. Les ventes de détail, reflet de la consommation, ont pour leur part chuté de 20,5% par rapport aux deux premiers mois de 2019
[2] L’artisanat, les petits commerces « non essentiels » et les professions libérales constituent le premier employeur de France
A écouter absolument https://www.youtube.com/watch?v=HSNJAcsygB0&feature=youtu.be
[3] « Je confirme les déclarations du chef d’état-major qui viennent de m’être rappelées par la direction centrale : il est absolument proscrit de porter le masque sur la voie publique ou à l’accueil du public. » Dans la bouche des policiers, il devient « interdit de se protéger ». « Le virus est dangereux pour le monde entier sauf pour les flics français ? », interroge légitimement l’un d’entre eux. Pour le moment, aucune réponse. Pendant ce temps-là, 100.000 masques vont être distribués en prison, difficile de comprendre. » Charlotte d’Ornellas 19/03/2020 Valeurs actuelles
[4] D’après le rapport de l’OMS 2020
[5] Élimination des insectes et animaux vecteurs, contrôle de l’eau potable et des eaux usées
[6] Nettoyage des vêtements et de la literie, hygiène corporelle, gestes barrières
[7] La peste noire, le choléra, le typhus, la typhoïde, la poliomyélite
[8] Data sourced from Japan Ministry of Health, Labour and Welfare and World Health Organization, as reported from 10:00|CET
[9] Jérémy André Comment j’ai été testé au coronavirus à Séoul Le Point 2 avril 2020 « Face à la pandémie, la Corée du Sud a évité le confinement généralisé. La clé : dépister, enquêter, isoler.
[10] Nombre de cas connus par 100000 habitants depuis le début de l’épidémie
[11] Au 19/3/2020 l’Italie est le pays qui totalise le plus grand nombre de décès : 3405 et la plus forte mortalité (nombre de décès / 100000 habitants) au monde.
[12] Un fils raconte avoir trouvé sa mère nue dans un sac à Rungis. S’en remettra -t-il ?
[13] Le port de masques et le respect de distance permettent de marcher ensemble sans risques.
[14] Fin que personne ne peut prévoir car les mesures adoptées qui étalent le pic de l’épidémie prolongent sa durée.
[15] Contrôle du SRAS et effets psychologiques de la quarantaine, Toronto, Canada Emerging Infectious Disease 2004 Juillet 10 (7) 1206 -1212
[16] D’après RFI les violences faîtes aux femmes en Argentine auraient augmenté de 60% depuis le confinement
[17] Ibid10
[18] L’Insee estime à 6 points de PIB l’impact d’un confinement de deux mois en France L’Obs du 26/3/2020
[19] « Coronaviré » : le chômage va exploser avec la crise L’Obs du 25/3/2020
[20] John P.A. Ioannidis A fiasco in the making? As the coronavirus pandemic takes hold, we are making decisions without reliable data March 17, 2020 https://www.statnews.com/2020/03/17/a-fiasco-in-the-making-as-the-coronavirus-pandemic-takes-hold-we-are-making-decisions-without-reliable-data/ John P.A. Ioannidis est professeur de médecine, d’épidémiologie et de santé des populations, de données de science biomédicale et de statistiques à l’Université de Stanford et codirecteur du Centre d’Innovation de Recherches Meta.
[21] Lors de l’épidémie de grippe H1N1 cette même équipe, avait prédit une scénario catastrophe avec plus de 8000 morts au Canada ; à la fin de l’épidémie les médecins canadiens en ont recensé 428.
[22] Delépine Quand les résultats de simulations mathématiques remplacent les résultats réels dans l’information Agoravox 19 10 2018 https://www.agoravox.fr/tribune-libre/article/quand-les-resultats-de-simulations-208775
[23] Au 10 avril 2020
[24] D’après l’OCDE sur 35 pays, la France se classe seulement au 19e rang ; lorsque le Japon bénéficie 7,8 de lits de ranimation, Taiwan de 7 et l’Allemagne de 6, la France n’en possède que 3 et l’Italie 2,6
[25] Le préfet de la région française la plus sévèrement touchée a même annoncé un plan de réduction de plusieurs centaines de personnel soignant.
[26] [15] https://psychologie-sociale.com/index.php/fr/experiences/influence-engagement-et-dissonance/204-la-soumission-a-l-autorite
[27] Conseil national de la résistance qui a mis en place la sécurité sociale
[28] « Le confinement est un fait inédit dans l’histoire de France, comme le rappelait Le Parisien. Même sous la botte hitlérienne, même sous le régime de Vichy, jamais les Français n’avaient été assignés à domicile du matin au soir. Dans le pays qui a le mot Liberté gravé sur tous ses frontons, pour aussi vertigineux que cela puisse paraître, c’est désormais chose faite. « Grâce » à Emmanuel Macron. » https://www.agoravox.fr/tribune-libre/article/confinement-de-macron-a-le-pen-222384
[29] Le gouvernement de Pétain appelait à la délation promue comportement civique.
[30] https://www.agoravox.fr/tribune-libre/article/confinement-de-macron-a-le-pen-222384
[31] Comme la durée hebdomadaire maximale du travail passée à 48 heures
[32] Nous allons bientôt accueillir les jeux olympiques avec nos sportifs dont la préparation a été gâchée par un confinement aveugle que n’ont pas subi ceux des pays plus démocratiques.
[33] « Les statistiques vont bon train, aux valeurs données dans l’absolu, sans aucune proportion avec les populations. On n’a par ailleurs, jamais donné autant de détails biologiques, épidémiologiques et étiologiques, concernant un microbe. Voilà que tout un chacun est médiatiquement confronté à ses propres ignorances scientifiques, ingénieriques-sociales comme cliniques. L’angoisse. Oui l’angoisse, d’autant plus que la veille, on regardait encore le sixième épisode de la série the Walking Dead ou the Rain sur Netflix, par exemple. L’époque en est là, de s’inventer des apocalypses zombies qui font de l’audimat » … https://www.agoravox.fr/culture-loisirs/parodie/article/cette-grippe-asiatique-222339
[34] L’OMS estime que la grippe saisonnière est chaque année responsable de plus de 4 millions d’infections et de plus de 500000 morts alors qu’au 10 avril 2020 le covid19 avait infecté 1 521 252 personnes et causé 92 798 morts.
[35] esp2017_5_principales_causes_de_deces_et_de_morbidite.pdf
[36]https://yetiblog.org/archives/22892?fbclid=IwAR0xkaM_pvxdzTMsR8sIQhLBAMPPlJB4KDvl6m7Ju3dGy5RTDfvPxTrACrc
[37] https://strategika.fr/2020/04/01/geopolitique-du-coronavirus-entretien-avec-valerie-bugault/
Géopolitique du coronavirus – entretien avec Valérie Bugault Strategika 1 avril 2020