FIL du diaporama 7 9 2019 Alix gardasil_ POUR SITE
Gardasil : le vaccin qui augmente le risque de cancer du col. Sera-t-il le prochain scandale sanitaire mondial ? par Gérard Delépine Chirurgien cancérologue gerard.delepine@bbox.fr
Indication officielle du Gardasil : prévenir le cancer du col de l’utérus en ciblant les virus papilloma humain. Est–ce en bonne voie ?
Pourquoi étudier l’efficacité des vaccins anti HPV contre le cancer du col ? Une propagande mondiale massive mensongère utilise la peur pour pousser les parents à vacciner leurs enfants, n’hésitant pas à les culpabiliser. La révélation des nombreuses complications ne ralentit pas l’hystérie vaccinale
Le president, Mme Buzyn et 50 sociétés savantes, syndicats et associations ~ sponsorisés par l’industrie ont proclamé leur volonté d’imposer la vaccination. En juillet 2018, 8 députés ont deposé un amendement de loi pour rendre cette vaccination obligatoire, et à nouveau sept autres en 2019.
DEFINITIONS
Quelques éléments démographiques sur le cancer du col. Ce cancer ne survient jamais avant 15 ans, est rarissime avant 20 ans et rare entre 20 et 25 ans. La vaccination a débuté en 2007, centrée sur les filles de 11-13 ans mais aussi « en rattrapage » celles de 13-18 ans et jusqu’à 26 ans en Australie.
En 2019, les registres du cancer ne donnent que les résultats de 2014-2016. On ne dispose donc que de 7 à 8 ans de recul analysables. Les filles qui avaient plus de 13 ans lors de la vaccination qui dépassent l’âge de 20 ans constituent le groupe témoin privilégié (20-24 ans)
L’histoire officielle du Gardasil est un beau conte qui repose sur 5 piliers : 1°) Les cancers du col de l’utérus, de la vulve, du vagin SERAIENT un cancer fréquent et une cause majeure de décès qui menace toutes les femmes. Vous et vos enfants seriez de plus exposés aux cancers de l’anus et ORL. 2°) Ces cancers SERAIENT dus à quelques souches de HPV (human papilloma virus) (Von Hauser récompensé d’un prix Nobel). 3°) GSK et Ian Frazer ont inventé des vaccins pour vaincre ces cancers.4°)Les essais auraient prouvé que les vaccins SERAIENT efficaces et bien tolérés. 5°) La vaccination devrait faire disparaître ces maux.
PREMIERE INTOX : problème majeur de santé publique? Faux en France! En France métropolitaine LE CANCER DU COL DE L UTERUS représente 1.7% des cancers avec une incidence de 6/100000, ce qui en fait une maladie rare (Une maladie est considérée comme rare par l’OMS lorsque son incidence est égale ou inférieure à 6/100000)
Aux USA, il ne figure même pas dans le graphique des cancers féminins du CDC, pourtant très pro gardasil) avec incidence de 7/100000 (contre 124/100000 pour cancer du sein).
En France,il est responsable de 0,6% de la mortalité par cancer avec une mortalité de 1,8/100000. Depuis la pratique des frottis moins de 1000 décès annuels lui sont imputables (contre > 5000 auparavant). Ces décès touchent majoritairement (70% à 80%) les femmes qui ne se font pas dépister selon les recommandations(1 fois / 3 ans à partir de 25 ans)
L’OMS, ministère et laboratoires pharmaceutiques utilisent la mauvaise situation sanitaire des pays sous développés pour créer la peur dans la population des pays riches (ceux qui peuvent payer ce vaccin hors de prix). La carte OMS montre qu’il n’est un problème de santé publique majeur uniquement dans les pays qui n’utilisent pas le dépistage par frottis.
Comment évaluer l’effet de la vaccination sur le risque de cancer invasif ? Pour évaluer réellement l’effet d’une action préventive de santé publique, il faut examiner l’évolution de l’incidence de la maladie dans la population générale et dans les groupes soumis à cette prévention. Et non pas utiliser des critères substitutifs (infection, anomalies cytologiques bénignes) dont la valeur n’a jamais été testée dans le cadre d’une vaccination et encore moins sur un échantillon de cobayes non représentatifs de la population concernée (essais thérapeutiques).
Pour un traitement censé prévenir le cancer du col, les registres officiels du cancer constituent les témoins indiscutables les plus pertinents.
Méthode pour évaluer objectivement l’effet de la vaccination sur le risque de cancer invasif : recueil des incidences du cancer du col de l’utérus rapportées dans les registres des cancers des pays qui utilisent le dépistage (pap tests) et dont la couverture vaccinale dépasse les 80%.
Analyse des évolutions avant et depuis la vaccination : en les comparant à celle de la France où la couverture vaccinale n’atteint pas 20%. Toutes courbes présentées ci après proviennent des instituts nationaux de statistiques (sauf pour l’Australie qui fournit des chiffres mais pas de courbe par groupe d’âge). Nous avons ajouté des commentaires en rouge. Voir toutes les courbes sur le diaporama.
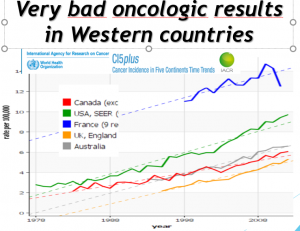
ANALYSE DES EVOLUTION DES PAYS QUI PRATIQUENT LE DEPISTAGE, UNE COUVERTURE VACCINALE >80%
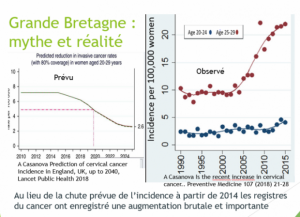
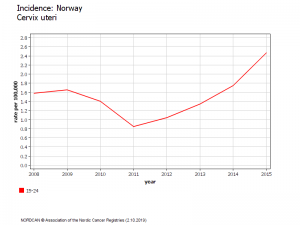
Période pré vaccinale (1980-2007) .L’incidence du cancer invasif du col a diminué spectaculairement dans tous les pays qui pratiquent le dépistage par frottis :le risque global de cancer invasif du col ne diminue plus dans les pays qui vaccinent, sauf pour les femmes de plus de 50 ans, non vaccinées, qui ont continué à bénéficier d’une baisse de leur risque de cancer.
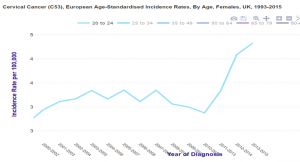
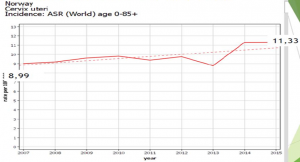
CHEZ LES FEMMES VACCINEES, AUGMENTATION IMPORTANTE et SIGNIFICATIVE DU NOMBRE DE CANCER DU COL 2 à 3 ans APRES VACCINATION
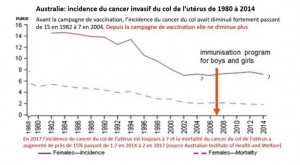
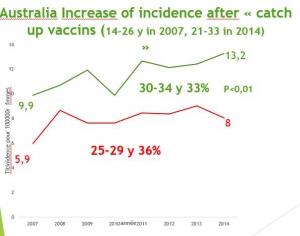
La politique vaccinale australienne. La campagne de vaccination a commencé en 2007 et a concerné les jeunes filles de 12 à 26 ans, qui avaient 20 à 34 ans en 2015 et représentent les groupes témoins pertinents.
Les mensonges publicitaires australiens. Les chiffres australiens officiels détaillés de cancers invasifs sont visibles sur les courbes du diaporama et contraires à la propagande médiatique qui se basent sur des simulations fondées sur des hypothèses érronées confondant infections et cancer .
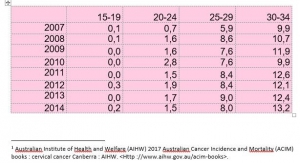
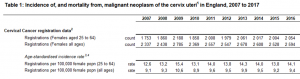
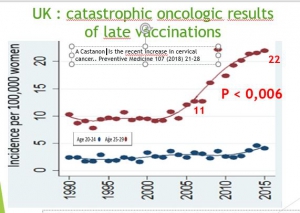
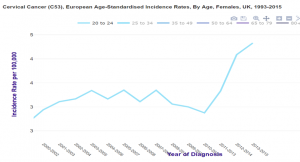
Grande Bretagne : augmentation d’incidence chez les 20-24 ans
Norvège : Evolution du risque chez les 20-24
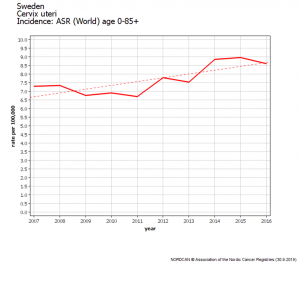
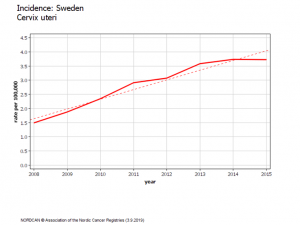
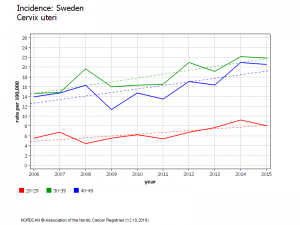
Suède évolution chez les 20-24 ans
Les filles âgées de 14 à 18 ans en 2010 ont subi des vaccinations de rattrapage pour 80% d’entre elles. Depuis, leur incidence (lissée sur trois ans) de cancer invasif du col de l’utérus a augmenté de 150%
Cette augmentation d’incidence est très significative et ne peut pas être due au hasard (P<0,001)
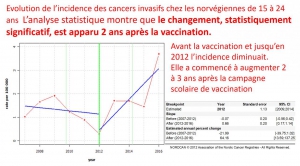
Recherche des points de rupture
La recherche des points de rupture permet d’établir la date de la modification de tendance evolutive et ainsi de vérifier s’il existe une corrélation temporelle entre celle-ci et la vaccination
En Suède, l’augmentation d’incidence est apparue deux ans après le début de la campagne scolaire de vaccination
USA : incidence du cancer invasif du col de l’utérus – 1975 à 2015
Depuis la vaccination, elle ne diminue plus
USA: incidence 2000-2016 chez les 15-39
Depuis 2013 l’incidence augmente chez les femmes jeunes
USA: tendance évolutive chez les américaines de plus de 40 ans
Durant cette même période, l’incidence a diminué chez les femmes de plus de 40 ans non vaccinées
Prédictions paradoxales de Nordcan, le registre scandinave
Au Danemark peu vacciné,Nordcan prédit un taux stable de cancer. En Suède, très vacciné, Nordcan prédit une augmentation importante du taux de cancer du col chez les vaccinées
Quelles sont les causes du cancer du col ?
FAKE NEWS : les virus seraient l’unique cause du cancer. Or le Papillomavirus est virus saprophyte. Toutes les femmes ont été un jour infectées par le HPV sans le savoir et ont guéri sans traitement.
Les HPV présents lors du cancer sont de simples témoins d’une activité sexuelle importante, source de microtraumatismes, de réinfections et d’inflammations répétés, causes possibles du terrain favorable aucancer
Infection n’ est pas cancer! Association ne veut pas dire causalité . L‘origine du cancer est multifactorielle . HPV sur les lieux du crime n’est qu’un témoin
Seule l’ analyse multifactorielle prenant en compte la totalité des facteurs connus précisant leur poids respectif aurait une réelle valeur évocatrice de causalité
Fake
les essais auraient prouvé vaccins efficaces et bien tolérés !Or la vaccination anti hpv est expérimentale
Depuis la commercialisation des vaccins : les études étiologiques se focalisent uniquement sur les papillomavirus !
Etat actuel des connaissances :
le papillomavirus pourrait n’être qu’un marqueur de risque de cancer traduisant son association avec les causes réelles (traumatismes répétés et inflammation chronique) …
L’évolution naturelle du cancer du col s’étalant sur deux décennies, on peut s’étonner de l’extrême précocité de l’augmentation de fréquence après vaccination.
Mais le cancer sous vaccins peut évoluer différemment. Ce n’est plus une évolution « naturelle »!
On le voit chaque jour avec les cancers soumis aux nouvelles drogues ciblées ou à l’immunothérapie à l’origine de cancers foudroyants quasi inconnus à cette échelle, avant l’ère de ces nouvelles molécules.
Pourquoi le Gardasil échapperait-il à cette possibilité de modification de l’histoire naturelle du cancer du col de l’utérus ?
Les vaccins peuvent modifier l’histoire des maladies
Les essais des premiers vaccins anti-sida Phambili puis Step ont abouti à une augmentation du risque d’attraper la maladie dans les années 2000 et ont été abandonnés pour cette raison
La vaccination anti hépatite B a été suivie dans les pays industrialisés (USA, France, GB, Australie, Canada) par une augmentation considérable du risque de de carcinomes hépatiques
La catastrophe toute récente du Dengvaxia contre la dengue aux Philippines* avec ses milliers de victimes (dont des dizaines de morts) est un autre exemple tragique.
Ces désastres sanitaires ont cruellement montré qu’une VACCINATION peut aggraver la maladie qu’elle était supposée prévenir.
Association ne veut pas dire causalité.
l’ origine du cancer est multifactorielle : précocité sexuelle, fréquence des rapports sexuels, le nombre de partenaires , la fréquence des infections bactériennes (chlamydia) ou virales HPV , Herpès, le tabagisme, le nombre de grossesses menées à terme, et même la cuisine au feu de bois.
Tous ces facteurs (souvent associés entre eux) sont statistiquement liés à un sur risque de cancer du col.
Fake
les essais auraient prouvé que les vaccins sont efficaces et bien tolérés.
Gardasil 4 antigènes souches type 6, 11, 16 et 18 et le Cervarix 2 souches (sur 150 souches connues)
infections 16 et 18 érigées en épouvantails par les industriels rares en Europe occidentale (étude en Catalogne)
le vaccin n’est actif que contre 1/2 infections à papillomavirus vues en France
Infection n’ est pas cancer
efficacité réelle sur infections et verrues génitales et prévention des anomalies CN2 et CN3, jamais démontrée dans la prévention du cancer invasif.
Aucune étude publiée ne prouve que le vaccin permet de diminuer le risque de cancer invasif ou le risque d’en mourir
La vaccination constitue donc une expérimentation.
notice du Gardasil : «Etant donné qu’aucun vaccin n’est efficace à 100 %, que Gardasil ne protège pas contre tous les types d’HPV ou contre des infections déjà existantes dues aux HPV, le dépistage en routine du cancer du col de l’utérus reste très important.. »
Vaccination expérimentale sur des populations entières Inutile (dépistage efficace)
Résultats actuels très inquiétants : dans tous les pays qui ont organisé des campagnes de vaccination, l’incidence des cancers du col a augmenté dans les groupes d’âge vaccinés. Notons que le vaccin anti HPV est hors de prix près de 250 euros pour 2 doses.
OMS : RECOMMANDE MAIS N EST PAS INDEPENDANTE !
Le budget de l’OMS est constitué pour 50% de contributions volontaires dont l’utilisation est décidée selon les désirs des donateurs…
Beaucoup de ces dons proviennent des firmes du médicament ou de fondations qui subordonnent leur attribution aux actions pro vaccinales (comme celles de Bill Gates) etc…
Corrélation obligations vaccinales/corruption
Les pays les plus corrompus sont ceux qui imposent les vaccinations. Informez-vous !