Gardasil alert, imminent risk of unnecessary and sometimes dangerous HPV vaccination for girls and boys.
Mon July 23, 2018 https://www.agoravox.fr/tribune-libre/article/gardasil-alerte-risque-imminent-d-206314#_ftnref25
By Gérard Delépine, MD, Orthopaedic Surgeon/Oncologist/statistician
English translation by Steve Hinks steve@hinksfamily.co.uk . In 2015 co-founded the UK Association of HPV Vaccine Injured Daughters (AHVID) share huge amounts of information obtained in particular by Freedom of Information Act requests and Parliamentary questions.
https://docteur.nicoledelepine.fr/
Open letter to parliamentarians, and to all citizens.
BE CAREFUL. While many doctors, foreign and French, citizens, patients sometimes victims, have been trying to inform for many years about the uselessness and the risks of the HPV vaccine, a new offensive of the pharmaceutical lobbies led again some MPs to try to introduce laws to make it a compulsory vaccination, already probably the most widespread in the world.
We have analysed the benefit-risk of this vaccine originally intended for women, but boys are likely to be targeted as well and denounced several times its uselessness coupled with its risks. both in women and in men. The time spent since FDA’s marketing authorization in June 2006 only adds new arguments against this vaccination, the strongest of which is the increase in the number of cervical cancers in the vaccinated population. which should encourage these countries to follow the example of Japan and Austria and to delete the recommendation.
Attention, some MPs want to impose a vaccination that can increase the risk of cervical cancer, as proven by international publications of national cancer registries.
ANALYSIS OF THE PROPOSED LAW WHICH MAY MAKE GARDASIL COMPULSORY
The preamble to the bill is based on the usual arguments of pharmaceutical companies widely disseminated by the media and their comfortably paid experts.
This preamble certainly recalls some indisputable true facts: there are more than one hundred HPV strains, the vaccines possibly protect against infection by the 4 to 9 strains included in the vaccine (only 2 to 5% of the 200 known strains ), against genital warts and some dysplasia’s without specifying that there is no evidence that it protects against cancer.
It is extremely disturbing to read in the presentation of opinion justifying the proposed law a number of known untruths:
« There are more than one hundred and twenty kinds of human papillomavirus (HPV), and fifteen are considered to be at high risk because they can cause cancers including HPV 16 and 18 causing 70% of infections. » But this only demonstrates a statistical correlation between presence of HPV and cancer, without anyone being so far able to demonstrate a direct CAUSALITY link.
« There are effective HPV vaccines. Current vaccines offer effective vaccination against 70% of carcinogenic HPV, and a new vaccine will soon increase this rate to 90%. » But, what do MPs mean by efficiency? the vaccine is effective on the infections of strains targeted by the vaccine (only 4 to 9 of the nearly 200 listed strains) but there is no evidence that it can prevent invasive cancer let alone avoid death by this cancer.
Citing Australia as a vaccine success story: « In Australia, where 80% of women and 75% of men are vaccinated, cases of HPV lesions have almost disappeared ». But, this statement is outrageous, as the following presentation will show you, because in this highly vaccinated country the number of cervical cancers (and other cancers attributed to HPV) continue to increase.
They also deny the risk of serious side effects that have led to protests in many countries (Denmark, Ireland, Japan, Colombia) and legal complaints from doctors against the EMA.
WHAT SHOULD IT EXPLAIN TO PARLIAMENTARIANS?
The regular smear (every three years) better guarantees early detection of cervical cancer.
In France, HPV infection is not a real public health problem in 2018, neither for women nor for men. In women, since smear screening has been used, the annual number of deaths from cervical cancer is consistently less than 1000 in France, and the women who die are almost exclusively those who have not been screened.
Diagnosis of HPV papillomavirus infection detected by systematic sampling should be avoided! Positive results often lead to unnecessary examination and very early conisation (biopsy) which is often useless.
The <1000 deaths per year from cervical cancer could all have been prevented by screening, Compare this with lung cancer (23,000 deaths), breast cancer (11,883 deaths), or prostate cancer (8,207 deaths) [15] .
Whilst efficacy of smear screening is proven, HPV vaccination not been proven to prevent a single invasive cervical cancer. The cancer registry records even suggest that this vaccine is sometimes likely to increase the risk.
INSTEAD OF REDUCING THE NUMBER OF CERVICAL CANCERS, IT INCREASES SOMETIMES
Curiously, the MPs who signed the bill do not talk about the proven results of the vaccine on the risk of invasive cancer of the cervix, its only official justification.
Instead of reducing the risk of invasive cancer of the cervix, HPV vaccines keep it at a high level or increase it!
After 12 years of use and more than 200 million girls vaccinated worldwide for a total bill of nearly $100 billion paid directly or indirectly by citizens , we can indeed draw a balance of effectiveness of vaccination in two ways:
1 °) By examining the evolution of the incidence (annual frequency of new cases per 100,000 women) of the invasive cancer of the cervix in each country, before and after vaccination, a method already validated in 2003.
2 °) World Standardised Rates: gross Incidence reported as « Standard World Population » to correct possible biases related to the demographic characteristics of each country
The evolution of the incidence of cervical cancer before and after vaccination with Gardasil can be traced in a perfectly reliable way in the national cancer registries controlled and published by the ministries of health of the concerned countries.
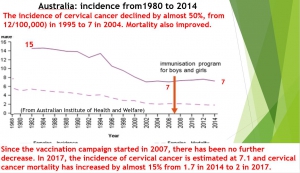
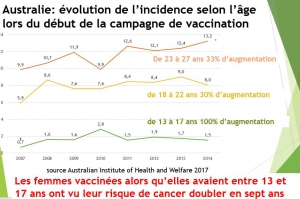
Australia, according to the Australian Institute of Health and Welfare, the incidence of cervical cancer declined by almost 50%, from 12(/100,000) in 1995 to 7 in 2004 (before the vaccination campaign). Mortality also improved, thanks to smear screening and treatment. However, since the vaccination campaign started in 2007, there has been no further decrease in either incidence or mortality. In 2017, the incidence of cervical cancer is estimated at 7.1 and cervical cancer mortality has increased by almost 15% from 1.7 in 2014 to 2 in 2017. And our MPs quote Australian efficiency!
The Australian Ministry of Health estimates the number of new cases of cervical cancer is 912 in 2017 and 930 in 2018. Claiming, like our MPs, that « cases of HPV lesions have almost disappeared » in Australia is therefore not correct. One cannot imagine that these MPs lie voluntarily, so we can conclude that they are poorly informed and that they should have checked themselves the information provided by the experts related to laboratories before distributing this « fake new ».
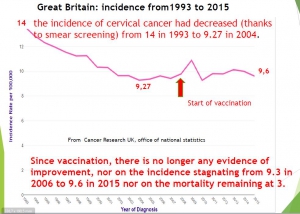
Great Britain, according to Cancer Research UK, the Office of National Statistics (ONS), the incidence of cervical cancer had decreased (thanks to smear screening) from 12.4 in 1995 to 9.27 in 2004. But since vaccination, there is no longer any evidence of improvement, nor on the incidence stagnating from 9.3 in 2006 to 9.6 in 2015 nor on the mortality remaining at 3.
Canada. According to the Canadian Cancer Society, the incidence of cervical cancer has decreased (through screening) from 18 in 1972 to 8.1 in 2008. But since vaccination, there has been no further progress on incidence stagnating at 8.3 in 2017.
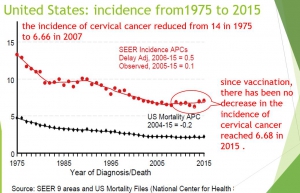
United States, according to the National Cancer Institute’s SEER cancer statistics review, the incidence of cervical cancer reduced from 14.8 in 1975 to 6.66 in 2007. But since vaccination, there has been no decrease in the incidence of cervical cancer reached 6.68 in 2015 .
Norway, according to the Cancer Registry of Norway, Oslo: before vaccination, the standardized incidence had fallen sharply thanks to smear screening from 24 in 1965 to 7 in 2004. But since the vaccination, it goes up to 13.9 in 2014 and 14.9 in 2015.
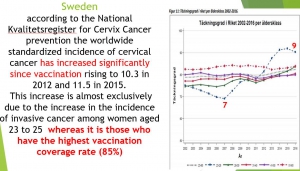
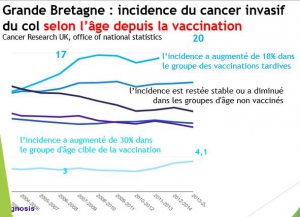
Sweden, according to the National Kvalitetsregister for Cervix Cancer prevention (NKCx ): before the vaccination campaign, the incidence of cervical cancer had decreased (thanks to screening) from 18 in 1967 to 7 in 2006. The worldwide standardized incidence of cervical cancer has increased significantly since vaccination rising to 10.3 in 2012 and 11.5 in 2015. This increase is almost exclusively due to the increase in the incidence of invasive cancer among women aged 23 to 49, which has reached more than 50% since 2006 (11 in 2006 versus 17 in 2015), whereas it is those who have the highest vaccination coverage rate (85%).
Thus, in countries whose populations have access to smear screening, it has led to a considerable reduction in the incidence of cervical cancer (from 40 to 60%). In contrast, the introduction of vaccination has not reduced the incidence or mortality of cervical cancer. Contrary to what is promised by laboratory-related physicians and by many global health authorities, vaccination campaigns have even been followed by an increase in the incidence of cancer.
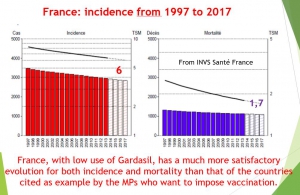
France, with low levels of HPV vaccination, can serve as a control country. According to Public Health France, the incidence of cervical cancer in mainland France has steadily decreased from 15 in 1995 to 7.5 in 2007, 6.7 in 2012 and 6 in 2017. This decrease in incidence was accompanied by a decrease in mortality from 5 in 1980 to 1.8 in 2012 and 1.7 in 2017. France, with low use of Gardasil, has a much more satisfactory evolution for both incidence and mortality than that of the countries cited as example by the MPs who want to impose vaccination.
Comparison of recent standardized incidences with vaccination coverage rates.
Immunization advocates claim that a high vaccination coverage rate reduces the risk of invasive cancer of the cervix. Yet the comparison of incidence and mortality rates with vaccine coverage rates shows the opposite:
Australia, HPV vaccination coverage exceeds 85% , but in 2017 the incidence of cervical cancer is 7 and the mortality is 2
Great Britain, despite vaccination coverage exceeding 80%, the incidence in 2015 was 9.6 and mortality 3
Sweden the vaccination coverage rate is close to 75% but the incidence 2015 reaches 11.5.
USA, in 2017 the vaccination coverage rate is 60% for a cervical cancer incidence of 6.8 and a specific mortality at 2.3.
France , in 2017, HPV vaccination coverage is very low (around 15% ) for a cervical cancer incidence of 6 and a specific mortality of 1.7
In countries with high immunization coverage, the incidence of invasive cancers and mortality are therefore higher than in France, and compulsory immunisation proposed by some MPs would eliminate this French paradox that protects our children!
The harmful side-effects are difficult to deny
In their preamble, the MPs deny that Gardasil can lead (as any treatment) to complications while Japan, Austria and Denmark have stopped promoting this vaccination after serious complications, sometimes even fatal and that families suffering from these vaccines organized public demonstrations in several countries of the world (Japan, Colombia, Ireland), and that Danish doctors lodged a complaint against the European Medicines Agency (EMA), which refused to answer the questions they asked. after the notification of severe neurological events not listed in the EMA registers.
In France, several lawsuits are in progress.
« Among the most frequently mentioned by the victims defended by M pathologies e Coubris include multiple sclerosis, lupus, disseminated acute encephalomyelitis (inflammation of the central nervous system) and myofasciitis macrophages (a disease that results in pain muscle and chronic fatigue) ». A parliamentary commission of inquiry which could hear experts, citizens and independent associations of laboratories, having, from near or far, no link of interest with laboratories, would be a first step to enlighten Parliament.
IN CONCLUSION, the benefit-risk balance is not in favour of vaccination, let alone compulsory.
A compulsory health measure should not be based on faith in vaccination or hidden conflicts of interest, but on proven facts, verifiable by every citizen. However, the facts established by the official records of cancer registries show that HPV vaccination does not protect against invasive cancer of the cervix but seems rather to maintain its frequency at a high level, and sometimes even increase it.
Let us fight against this bill that threatens our children, by informing everyone, our MP, our senator, our elected officials, that no one may be unaware.
Only this work of proximity of each citizen this summer, will be able to avoid this new catastrophe of return which could be the anti-HPV vaccination, as it has been and still is the obligation of vaccination against hepatitis B.
Let’s apply the precautionary principle! Let us respect the right of every human being to informed choice/consent for medical interventions!