PARADOXICAL EFFECT OF ANTI-HPV VACCINE GARDASIL ON CERVICAL CANCER RATE
State of published results in registers, on January 2019
Dr G Delépine, oncologist, surgeon
gerard.delepine@bbox.fr
“How wonderful that we have met with a paradox. Now we have some hope of making progress”. Niels Bohr (Nobel prized for his works on the structure of the atom and chemical reactions )
Changing the natural history of cancer that increases in frequency and occurs faster.
It takes a long time to affirm that a preventive action really protects. But the failure of this supposed protection can sometimes be very quickly obvious. To prove that the Titanic was truly unsinkable would have required decades of navigation on the most dangerous seas of the world. Demonstrating that it wasn’t, took only a few hours … This » Titanic » demonstration is unfortunately reproduced by the Gardasil vaccination.
Evidence that vaccination increases the risk of invasive cancer can be rapid, if the vaccine changes the natural history of cancer by accelerating it. The analysis of trends in the incidence of invasive cervical cancer published in official statistics (registers) was studied in the first and most fully vaccinated countries (Australia, Great Britain, Sweden and Norway). Unfortunately, it’s the case for HPV vaccines.
Pre-vaccination period : spectacular success of cervical smear screening with a steady decrease in the rate of invasive cervical cancer.
In all countries that performed smear screening, the pre-vaccination period from 1989 to 2007 was marked by a significant decrease in the standardized incidence of cervical cancer.
In less than 20 years, the incidence of invasive cancer of the cervix decreased from :
13.5 to 9.4 n Great Britain [1]
13.5 to 7 in Australia[2] ,
11.6 to 10.2 in Sweden [3],
15.1 to 11 in Norway [4],
10.7 to 6.67 in the USA [5],
11 to 7.1 in France.
Globally, in the countries that used smear screening, the average annual rate of decline was 2.5% between 1989 and 2000 and 1% between 2000 and 2007, resulting in a total decrease of nearly 30% across 1989-2007.
Era of vaccination: reversal of the trend. Gardasil’s prevention failure erases the beneficial effects of the smear and accelerates the onset of cervical cancer.
Since vaccination, in all the countries implemented with a large vaccination program, there is a reversal of the trend, with a significant increase in the frequency of invasive cancers in the most vaccinated groups. Let’s look at OFFICIAL sources.
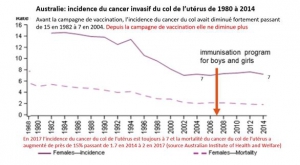
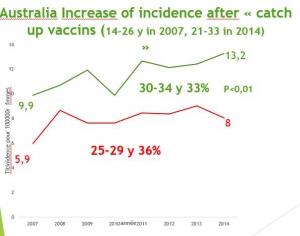
AUSTRALIA : contrary to the FAKE NEWS OF THE MEDIA AND POLITICS, REGISTER DOES NOT SHOW CANCERS OF THE CERVIX DISAPPEAR, BUT INCREASE.
Australia was the first country to organize routine immunization for girls (April 2007 school-based program for females aged 12–13 years, July 2007 time-limited catch-up program targeting females aged 14–26 years) and then for boys (2013).. According to the last Australian Institute of Health and Welfare publication (2018 publication describing the detailed rates until 2014 ) [6] , the standardized incidence in the overall population has not decreased since vaccination 7/100000 in 2007 versus 7.4 in 2014.
This global stabilization results from two contradictory trends that only appears by examining trends, according to age groups.
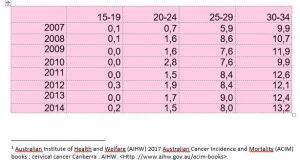
Vaccinated age groups women have seen their risk increase:
100% increase for those aged 15 to 19 (from 0.1 in 2007 to 0.2 in 2014)
113% increase (from 0.7 to 1.5) in groups aged 20 to 24 more than 80% of them were catch up vaccinated when 13 to 17 years old.
But, as the figures are very small, this increase does not reach statistical significance.
About a third increase for 25-29 group (from 5.9 to 8 ,p=0.06) and for 30-34 (from 9.9 to 12.4 c=0.80 p=0.01) less vaccinated. These increases are statistically significant cannot be due to hazard.
A drama known to one top athlete : Sarah Tait
This increased risk of cancer following vaccination was dramatically illustrated by the sad story of Sarah Tait, olympic rowing champion, at the 2012 London Olympics. This champion saw her life shattered in full glory : she suffered invasive cervical cancer a few years later, being vaccinated and died at age 33. Of course, we don’t know if vaccination was the direct cause of her cancer, but she has, statistically, a one in two chances of having suffered from a cancer linked to vaccination (to be part of the 113% increase of cancer observed after vaccination). In addition, we remark that cancer appears very early in this woman.
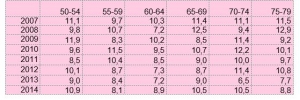
Non vaccinated women continue to benefit from screening with pap smear
During the same period, older women (and therefore unvaccinated) saw their cancer risk decrease significantly:
less 17% for women aged 55 to 59 (from 9.7 to 8.1),
less 13% for women aged 60 to 64 ( from 10.3 to 8.9),
less 23% for those aged 75 to 79 (from 11.5 to 8.8)
and even less 31% for those aged 80 to 84 (from 14.5 to 10).
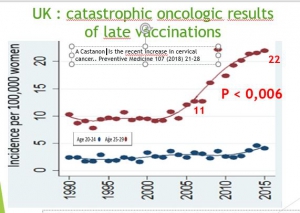
GREAT BRITAIN : THE PARADOXICAL EFFECT OF GARDASIL PROMOTING CANCER
In UK, a national program was introduced in 2008 to offer HPV vaccination routinely to 12–13-year-old and offer catch-up vaccination to girls up to 18 years old. The UK national program initially used the bivalent HPV vaccine (Cervarix), but, changed in 2012 to use the quadrivalent vaccine (Gardasil). HPV vaccination coverage in England has been high with over 80% of 12–13 years old receiving the full course coverage. The catch-up cohort has been lower covered (ranging from 39% to 76%).
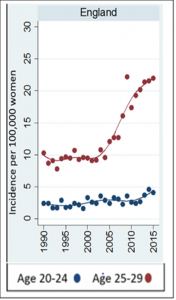
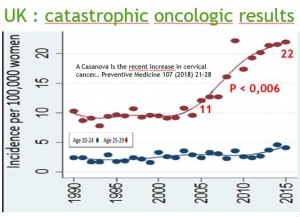
Since the vaccination, the standardized incidence in the overall population increased from 9.4 per 100000 in 2007 to 9.6 in 2015. We observe contrasting trends between the age groups.
Vaccination promoters expected cervical cancer rates decrease in women aged 20 to 24 from 2014, as vaccinated adolescents enter their second decade. However, in 2016, national statistics showed a sharp and significant increase in the rate of cervical cancer in this age group. This information of 2016 has unfortunately not been publicized. They could have served as an alert.
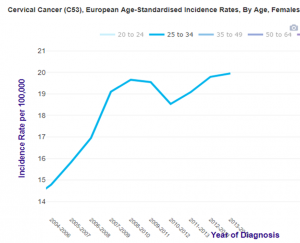
Women aged between 20 and 25 years, vaccinated for more than 85% of them, when they were between 14 and 18 years old, have seen their cancer risk increase by 70% in 2 years (from 2.7 in 2012 to 4.6 per 100,000 in 2014 p = 0.0006) and those aged 25 to 30, ( aged between 18 and 23 at the time of the vaccination campaign) have seen their cancer risk increase by 100% between 2007 and 2015[7] (from 11 / 100,000 to 22 / 100,000 ).
Women 25 to 34 years, (less vaccinated, only exposed to some catch-up vaccinations), have seen their risk increased by 18% (from 17 in 2007 to 20 in 2014).
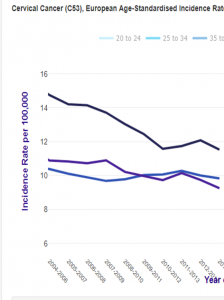
In Great Britain, as in Australia, older, unvaccinated women have seen their risk decrease:
( -13% for women aged 65 to 79 and -10% for those over 80 ), most likely because continuation of smear screening.
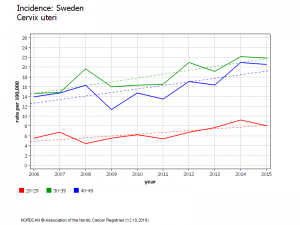
SAME PARADOXICAL PHENOMENON OF GARDASIL IN SWEDEN : THE RATE OF CANCER INCREASES IN THE VACCINATED AGE GROUPS . ALERT!
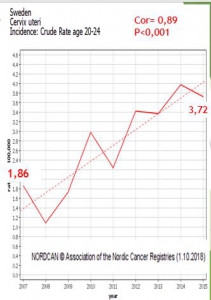
In Sweden, Gardasil has been used since 2006. The vaccination program was rolled out in 2010, with vaccination coverage of 12-year-old girls approaching 80%. In 2012-2013, with a catch-up program, almost all girls aged 13 to 18 were vaccinated.
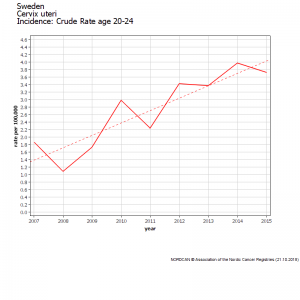
In this country, the standardized incidence of cervical cancer in the global population has increased steadily since vaccination from 9.6 per 100000 in 2006 to 9.7 in 2009, 10.3 in 2012 and 11.49 in 2015[8]. This increase is mostly due to the increase in the incidence of invasive cancers among women aged 20-24 whose incidence doubled ( from 1.86 in 2007 to3.72 in 2015 p<0.001)[9] and in women aged 20 to 29 the incidence of invasive cancer of the cervix increased by 19% (from 6.69 to 8.01)
In contrast, as in Australia and Great Britain, a decrease in the incidence of invasive cancer has been observed in women over 50, a group that has not been included in the vaccination program. The incidence of invasive cancer of the cervix decreased between 2007 and 2015 by 6% for women aged 50 to 59 (from 14.24 to 13.34), and 4% for those aged 60 to 69 (12.63%). at 12.04,) 17% for those aged 70 to 79 (from 15.28 to 12.66) and 12% for those over 80 (from 15.6 to 13.68).
IN NORWAY
Cancer registry shows an increase in the standardized incidence of invasive cancer of the cervix from 11.7 in 2007 to 12.2 in 2009, 13.2 in 2012 and 14. 9 2015 [10].
This increase is due -almost exclusively- to young women, which include all vaccinated, as evidenced by the sharp decline of the average age of onset of the cervix cancer from 48 years in 2002 -2006 to 45 years in 2012-2016.
Between 2007 and 2015 , the incidence of invasive cervical cancer increased by 8% among women aged 20 to 29 (from 7.78 to 8.47)[11].
During the same period, a decrease in the incidence of invasive cancer was observed in older women, not involved in the vaccination program: -11% for women aged 55 to 64 (15.47 to 13.7), -16% for those aged 65 to 74 (17.7 to 14.71) and -29% for those aged 75 to 85 (18.39 to 13) .
In USA
In this country, vaccination coverage is lower than in previous countries (close to 60%).
According to the Cancer Statistics Review 1975-2015[12], the standardized incidence of invasive cervical cancer remains stable(+0.1) since vaccination.
In US, the same discrepancy is observed according to age groups, but of lesser amplitude. Women over 50, benefit a 5% decrease in their risk (from 10.37 per 100000 in 2007 to 9.87 in 2015), whereas younger women, which include vaccinated, have given their risk increase of 4% (5.24 in 2007 to 5.47 in 2015).
WITNESS COUNTRY: FRANCE
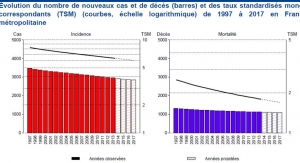
The evolution of these countries, with high immunization coverage, can be compared to the trend observed in metropolitan France, where HPV vaccination coverage is very low (around 15%). France can be considered, for this reason, as a control country. In France[13] the incidence of cervical cancer has steadily decreased from 15 in 1995 to 7.5 in 2007, 6.7 in 2012 and 6 in 2017, much lower than those of countries with high vaccine coverage.
This decrease in incidence was accompanied by a decrease in mortality from 5 in 1980 to 1.8 in 2012 and 1.7 in 2017.
It is paradoxical and very worrying that these excellent French results, with low cervix cancer rate and low related mortality, could be jeopardized by an obligation considered in the short term by our policies, for some misinformed and other big pharma links[14].
DRAMATIC AND UNEXPECTED PARADOXICAL EFFECT OF GARDASIL: THE ALERT MUST BE GIVEN TO DECISION MAKERS AND THE MEDIA.
In all countries that achieved high HPV vaccination coverage, official cancer registries show an increase in the incidence of invasive cervical cancer.
For women under 20, the crude number are to small to reach statistical significance, but the similar increases in all the studied countries constitutes a strong alarm signal.
For women 20-30 the incidence increases after catch up vaccination, and is highly significant (p<0.01or 0.001). In these same countries, during the same period, older women, not vaccinated, have seen their risk of cervical cancer continue to decline.
Similarly, in metropolitan France, a country with low vaccination coverage, the incidence of cervical cancer continues to decline at a rate comparable to the pre-vaccination period.
These paradoxical results plea for a rapid revision of recommendations and intensive research to explain this catastrophic issue.
[1] Cancer Research UK, Cervical Cancer (C53): 1993-2015, European Age-Standardized Incidence Rates per 100,000 Population, Females, UK Accessed 08 [ 2018 ].
[2] AIHW [2]. 13. AIHW 2017. Cancer in Australia 2017. Cancer series no. 101. Cat. No. CAN 100. Canberra: AIHW.
[3] NORDCAN, Association of the Nordic Cancer Registries 3.1.2018
[4] Bo T Hansen, Suzanne Campbell, Mari Nygård Long-term incidence of HPVrelated cancers, and cases preventableby HPV vaccination: a registry-based study in Norway BMJ Open 2018; 8: e019005
[5] Table 5.1 Cancer of the Cervix Uteri (Invasive) Trends in SEER Incidence and US Mortality SEER Cancer Statistics Review 1975-2012
[6] Australian Institute of Health and Welfare (AIHW) 2017 Australian Cancer Incidence and Mortality (ACIM) books: cervical cancer Canberra: AIHW. <Http://www.aihw.gov.au/acim-books>.
[7] A Castanona, P Sasienia Is the recent increase in cervical cancer in women aged 20-24 years in
England a cause for concern? Preventive Medicine 107 (2018) 21-28
[8] Nationellt Kvalitetsregister für Cervix cancer prevention (NKCx), http://nkcx.se/templates/_rsrapport_2017.pdf [in Swedish]
[9] Engholm G, Ferlay J, Christensen N, Hansen HL, Hertzum-Larsen R, Johannesen TB, Kejs AMT, Khan S, Olafsdottir E, Petersen T, Schmidt LKH, Virtanen A and Storm HH: Cancer Incidence, Mortality, Prevalence and Survival in the Nordic Countries, Version 8.1 (28.06.2018). Association of the Nordic Cancer Registries. Danish Cancer Society. Available from http://www.ancr.nu, accessed it 30 / 09 / 2018 .
[10] Cancer in Norway 2016
[11] Engholm G, Ferlay J, Christensen N, Hansen HL, Hertzum-Larsen R, Johannesen TB, Kejs AMT, Khan S, Olafsdottir E, Petersen T, Schmidt LKH, Virtanen A and Storm HH: Cancer Incidence, Mortality, Prevalence and Survival in the Nordic Countries, Version 8.1 (28.06.2018). Association of the Nordic Cancer Registries. Danish Cancer Society. Available from http://www.ancr.nu, accessed is 1 / 10 / 2018
[12] SEER 9 National Center for Health Statistics, CDC
[13] Francim, HCL, Public Health France, INCa. Projections of Cancer Incidence and Mortality in Metropolitan France in 2017 – Solid Tumors [Internet]. Saint-Maurice: Public health France [updated 02/01/2018; viewed on the 09/05/2018
[14] https://www.agoravox.fr/tribune-libre/article/gardasil-alerte-risque-imminent-d-206314 Gardasil, alert, imminent risk of mandatory vaccination against HPV unnecessary, and sometimes dangerous , for girls and boys.